2020
UPR을 활용하기 위한 소분자 전략: 여기에서 어디로 가야 할까요?
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7667976/
(UPR)은 병리학적 ER 스트레스에 대한 반응으로 소포체(ER) 및 전체 세포 생리학을 조절하는 데 중심 역할을 합니다.
UPR은 ER 막 단백질 IRE1, ATF6 및 PERK의 다운스트림에서 활성화된 세 가지 신호 전달 경로로 구성됩니다.
일단 활성화되면, 이들 단백질은 ER 스트레스를 완화하고, 세포 생리를 적응시키며, 세포 운명을 결정하는 기능을 하는 전사 및 번역 신호를 개시합니다.
UPR 신호전달의 불균형은 많은 신경퇴행성 질환, 단백질 접힘 질환, 당뇨병, 허혈성 장애 및 암을 비롯한 수많은 병인학적으로 다양한 질병의 발병기전과 관련이 있습니다.
이것은 IRE1, ATF6, 또는 PERK 신호 전달은 이러한 다양한 질병과 관련된 UPR 신호 전달의 병리학적 불균형을 개선하고 다양한 세포 및 유기체 맥락에서 UPR의 중요성을 정의합니다.
소포체(ER)는 단백질 분비, 지질 합성 및 칼슘 조절을 포함한 중요한 생물학적 기능과 관련이 있습니다.
따라서 끊임없이 변화하는 생리적 환경에 대응하여 ER 기능을 적절하게 조절하는 것은 유기체의 생존에 매우 중요합니다.
질병의 맥락에서 ER 기능을 변경하는 가장 매력적인 전략 중 하나는 ER 손상( 즉, ER 스트레스) 후 ER 및 세포 생리를 조절하는 주된 스트레스 반응 신호 경로인 UPR(Unfolded protein response)을 표적으로 삼는 것입니다.
UPR은 ER stress-sensing transmembrane protein inositol-required enzyme 1(IRE1),
protein kinase R-like endoplasmic reticulum kinase(PERK),
그리고 Activate transcription factor 6(ATF6)의 3가지 신호전달 경로로 구성되어 있습니다.그림 1).
이 세 가지 신호경로는 ER 내강 내 비천연 단백질 축적 및 ER 막 내 지질 불균형을 포함하여 다양한 유형의 ER 스트레스에 대한 반응으로 활성화됩니다.
이러한 UPR 경로의 활성화는 ER 스트레스를 완화하고 급성 손상 후 세포 적응을 촉진하는 기능을 하는 ER 및 전체 세포 생리학의 전사 및 번역 리모델링을 이끌어냅니다.그림 1).
이 활동을 통해 UPR은 분비성 단백질 항상성 유지, 증식, 산화환원 조절, 분화 및 대사를 포함하여 세포 생리학의 다양한 측면을 조절하는 데 관여하는 보호 신호 전달 경로로 기능합니다.
그러나 보호 리모델링을 통해 완화할 수 없는 만성 또는 심각한 ER 손상에 대한 응답으로 연장된 UPR 활성화는 세포자멸사 신호 전달을 유도합니다.
따라서 UPR은 병리학적 ER 스트레스에 대한 반응으로 보호 및 세포 사멸 신호를 지시하는 데 이러한 두가지 현상의 전부에 중요한 역할을합니다.
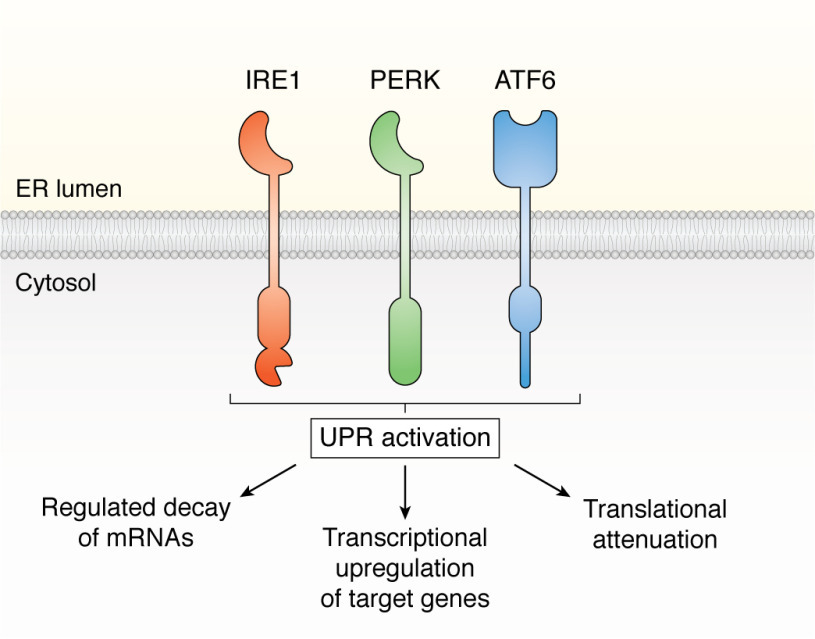
UPR 신호를 활성화하는 세 가지 ER 스트레스 감지 단백질.
IRE1, PERK 및 ATF6의 활성화는 번역 및 전사적으로 ER 및 세포성 단백질 유지를 리모델링하는 통합 신호 전달을 촉진합니다.
ER 기능을 조절하기 위한 UPR 신호전달의 중요성으로 인해 UPR 신호전달의 변경이 인간 질병 발병에 기여한다는 것은 놀라운 일이 아닙니다.
UPR 신호 전달의 환경적 또는 노화 관련 결핍은 심혈관 질환 및 신경퇴행성 질환을 포함한 다양한 유형의 질병에 기여합니다.
대조적으로, UPR 신호의 과활성은 또한 질병 발병과 관련이 있습니다.
예를 들어, 과활성 PERK 신호 전달은 다양한 신경퇴행성 질환과 관련이 있습니다.
유사하게, 만성 IRE1 활성은 마우스 모델에서 죽상동맥경화증과 관련이 있습니다.
따라서 UPR 신호 전달 경로를 통한 신호 전달이 너무 많 거나 너무 적으면 인간 질병의 맥락에서 발병을 촉진할 수 있습니다.
병인학적으로 다양한 질병의 발병기전에서 변경된 UPR 신호전달의 중요성은 이러한 경로를 치료 개입의 매력적인 표적으로 만듭니다.
UPR의 IRE1
IRE1 신호전달 경로는 효모에서 인간에 이르는 모든 유기체에서 발견되는 UPR의 가장 고도로 보존된 부분입니다.그림 1).
특히, 식별된 최초의 UPR 경로였으며 가장 잘 연구된 경로일 것입니다.
IRE1은 ER 내강 도메인, 세포질 키나제 도메인 및 세포질 RNase 도메인의 세 가지 도메인을 포함하는 I형 ER 막 단백질입니다.그림 2, A 및 B ).
포유류는 IRE1α 및 IRE1β의 두 가지 다른 IRE1 동형을 인코딩합니다.
IRE1α는 대부분의 세포 유형에서 UPR 신호 전달과 관련된 주요 동형인 반면, IRE1β는 주로 조직 특이적 메커니즘 및/또는 IRE1α 활성 조절을 통해 기능하는 것으로 보입니다.
이 검토의 목적을 위해 우리는 주로 IRE1α 동형체(달리 언급되지 않는 한 여기에서는 IRE1이라고 함)에 중점을 둡니다.

IRE1 UPR 신호 전달 경로의 약리학적 표적.
A , ER 스트레스 의존적 활성화 및 IRE1 UPR 신호 전달 경로의 다운스트림 신호 전달의 단순화된 메커니즘.
B , 내강 도메인, 막횡단 도메인( TM ), 세포질 키나제 및 RNase 도메인을 포함하는 IRE1의 도메인 구조 .
주요 도메인의 효소 활성은 A에 표시된 대로 IRE1의 세포질 키나제 도메인이 트랜스 자가인산화에 참여하고 활성화 시 RNase 도메인은 XBP1 mRNA 스플라이싱 및 RIDD를 통해 기능합니다 .
C , 인간 IRE1의 뉴클레오티드-결합 포켓에 대한 ADP의 결합을 보여주는 이미지.
D, IRE1 RNase 활성 부위 억제제 4μ8c 및 STF-083010의 구조.
E , IRE1 키나제 활성 및 RNase 활성 둘 다를 억제하는 유형 II IRE1 키나제 억제제 화합물 3 및 KIRA6의 구조.
F , IRE1 RNase를 알로스테릭하게 활성화하면서 IRE1 키나제 활성을 억제하는 I형 IRE1 키나제 억제제의 구조.
G , 전사 프로파일링을 우선시하는 HTS를 통해 확인된 새로운 IRE1/XBP1s 활성화 화합물 IXA1, IXA4 및 IXA6의 구조.
IRE1은 ER 스트레스 및 지질 불균형을 포함한 다양한 세포 손상에 대한 반응으로 활성화됩니다.
UPR의 가장 잘 연구된 분야임에도 불구하고 IRE1 활성화의 분자 메커니즘은 IRE1 내강 도메인에 의한 ER 스트레스 감지를 포함하는 여러 제안된 모델과 함께 다소 논란의 여지가 있습니다.
한 저명한 모델은 IRE1이 (ERdj4)와 같은 BiP 보조 샤페론에 의해 조절되는 과정을 통해 단백질(BiP)과 IRE1 내강 도메인 사이의 동적 상호작용을 통해 ER 스트레스를 감지한다고 제안합니다.
이 모델에서 BiP는 잘못 접힌 단백질이 ER 내강과 함께 축적됨에 따라 IRE1 내강 도메인에서 분리되어 이 경로를 통한 신호 전달을 활성화합니다.그림 2).
그러나 다른 모델은 IRE1이 IRE1 내강 도메인에서 발견되는 추정되는 펩티드 결합 홈에서 비천연 단백질 구조에 직접 결합한다고 제안하며, 이는 IRE1이 ER 스트레스 동안 비천연 단백질의 축적을 직접 감지한다는 것을 시사합니다.그림 2A ) .
IRE1 활성화의 정확한 메카니즘은 여전히 조사되고 있지만 IRE1 신호전달의 다운스트림 이벤트는 IRE1 올리고머화, 자가인산화 및 RNase 활성화를 포함하는 것으로 잘 확립되어 있습니다.그림 2가 ).
IRE1은 주로 소포체 막에 단량체로 존재하며 소포체 스트레스에 대한 반응으로 이량체화/올리고머화 및 후속적인 트랜스 자가인산화(trans-autophosphorylation)를 겪습니다.그림 2A ) .
IRE1 자가인산화는 세포질 키나제 도메인 내의 보존된 "활성화 루프"의 여러 부위에서 발생합니다.그림 2다 )
RNase 도메인은 X-box 결합 단백질 1( XBP1)을 인코딩하는 mRNA를 절단 한 다음 RTCB RNA 리가아제에 의해 다시 연결되어 이 전사체에서 프레임 이동을 유발합니다.그림 2A )
일반적으로, XBP1s 전사 신호는 급성 스트레스에 대한 반응으로 ER 단백질 유지 및 세포 적응을 촉진하는 보호 메커니즘입니다.
XBP1의 활성화를 통해 전사 변화를 유도하는 것 외에도 IRE1은 ER 스트레스에 따른 스트레스 반응 신호 전달에서 역할을 하는 다른 기능을 가지고 있습니다.
이러한 추가 기능 중 하나는 활성화된 IRE1 RNase가 다양한 ER 관련 mRNA를 분해하는 조절된 IRE1 의존성 붕괴(RIDD)라고 합니다.그림 2A )
보호 XBP1s 전사 신호와 달리 IRE1 RIDD 활성은 보호 신호와 세포자멸사 신호 전달에 모두 관련되어 있습니다.
예를 들어, RIDD 활성은 ER 내에서 유입되는 단백질 접힘 부하를 감소시키고 사멸 수용체 5( DR5 )를 인코딩하는 mRNA의 분해를 통해 세포자멸사 신호전달을 억제합니다
따라서, RIDD 활성은 심각하거나 장기간의 ER 스트레스에 대한 반응으로 IRE1 신호전달이 적응성에서 친-세포자멸사로 전환되는 데 관여하는 것으로 보입니다.
IRE1은 또한 RNase 활성과 무관한 신호 전달을 촉진합니다. 활성 인산화된 IRE1은 종양 괴사 인자 수용체 관련 인자 2(TRAF2)에 결합하여 ASK-JNK 신호 전달 축의 하류에서 세포자멸사 신호를 촉진하고 핵 인자 κB(NFκB)의 하류에서 염증 신호를 촉진할 수 있습니다.그림 2A )
이 IRE1-TRAF2 신호는 지방간 질환 및 신경변성을 비롯한 병리학적 상태와 관련된 중증 또는 만성 ER 손상에 대한 반응으로 세포 사멸 및 염증을 촉진합니다
RNase 억제제로 IRE1 신호 전달 방지
IRE1 활성은 종종 급성 ER 스트레스 동안 보호적인 반면, 이 경로의 만성 활성은 수많은 질병 병인과 관련되어 있으며 특정 암의 지속성을 지원할 수 있습니다.
따라서, IRE1 신호전달의 강력한 억제제를 개발하기 위해 여러 전략이 사용되었습니다. 그러한 전략 중 하나는 XBP1 스플라이싱 을 방지하기 위해 IRE1 RNase 활성을 차단하는 화합물 개발에 중점을 둡니다 .
중요하게도, IRE1 RNase 억제제의 사용은 과민성 IRE1 신호 전달과 관련된 질병 발병에 대응하는 잠재적인 치료 전략으로서의 가능성을 보여주었습니다.
쥐의 고지혈증에 대한 반응으로 IRE1의 하류에 있는 염증 인자 IL-1β와 IL-18의 전신적인 상향 조절을 방지합니다.
죽상동맥경화증의 마우스 모델에서 면역 반응과 죽상경화반 발달을 낮춥니다
다발성 골수종 모델에서 암세포 증식을 늦추어 화학요법의 한 방식으로 발암성 세포에서 생존 UPR 기능을 억제할 가능성을 보여줍니다
중요한 것은 4µ8c와 STF-083010이 모두 널리 사용되고 제한된 독성을 나타내는 반면 4µ8c는 일부 보고된 표적 외 효과가 있다는 것입니다.
예를 들어, 췌장 β 세포를 4μ8c로 처리하면 IRE1 RNase 억제와 무관하게 인슐린 분비가 감소했습니다
또한, 4μ8c는 항산화 특성을 갖는 것으로 보이며, 이는 안지오텐신 II에 의해 유도된 활성 산소 종 생성 감소로 입증됩니다
키나제 억제제로 IRE1 자가인산화 억제
키나제 억제제는 반대 형태의 키나제 활성 부위를 안정화시키는 능력에 따라 유형 I 또는 유형 II로 분류됩니다.
IRE1과 관련하여 II형 억제제는 IRE1 자가인산화와 IRE1 RNase의 알로스테릭 활성화를 모두 차단하는 형태로 IRE1 활성화 루프를 안정화시키는 것으로 나타났습니다
키나제 억제제를 사용한 IRE1 RNase 활성의 알로스테릭 활성화
유형 I 키나제 억제제는 IRE1 키나제 활성 부위 내의 활성화 루프 형태에 대해 유형 II 억제제와 반대되는 효과를 나타냅니다.
따라서 유형 I 키나제 억제제는 IRE1 자가인산화를 억제하면서 IRE1 RNase 도메인을 알로스테릭하게 활성화할 것으로 예측됩니다.
중요하게도, 세포 기반 연구는 낮은 마이크로몰 농도에서 APY29 치료로부터 다면발현성 독성을 나타내어 이 화합물을 다양한 세포 상황에 적용하기 어렵게 만듭니다.
유형 I 및 유형 II IRE1 키나제 억제제는 IRE1 활성화의 분자 메커니즘을 해체하는 데 매우 유용한 것으로 입증되었으며 RNase 도메인을 직접 표적화하지 않고 키나제 억제를 통해 IRE1 RNase 신호를 억제하거나 활성화할 수 있는 독특한 기회를 허용합니다.
그러나 이러한 화합물의 표적을 벗어난 활성(다른 키나제에 대한 결합과 관련이 있을 수 있음), 독성 및/또는 낮은 생체이용률로 인해 이러한 화합물은 복잡한 세포의 맥락에서 IRE1 신호전달의 기능적 의미를 결정하는 데 덜 유용함이 입증되었습니다.
UPR의 PERK
PERK UPR 신호 전달 경로는 약리학적 개입을 위한 매우 매력적인 표적임이 입증되었습니다.
PERK는 N-말단 루미날 도메인과 세포질 이펙터 키나제 도메인으로 구성됩니다(그림 3, A 및 B ).
IRE1과 마찬가지로 PERK는 ER 스트레스와 지질 불균형에 의해 활성화될 수 있습니다.
흥미롭게도 PERK 활성화 메커니즘은 동일하지는 않지만 IRE1에서 관찰된 메커니즘과 유사합니다.
ER 스트레스에 대한 반응으로 BiP는 PERK 내강 도메인에서 해리되어 세포질 PERK 키나제 도메인의 올리고머화 및 자가인산화를 촉진합니다.그림 3A ).
그러나 IRE1과 달리 ERdj4와 같은 BiP 보조 샤페론은 PERK 신호 조절에 관여하지 않는 것으로 보이며, 이러한 다양한 ER 스트레스 센서의 활성화 사이의 미묘한 차이를 강조합니다.
일단 활성화되면, 결과적으로 리보솜 단백질 합성이 전체적으로 감소합니다.
이 감소는 ER 내강으로 들어가는 새로 합성되고 펼쳐진 단백질의 부하를 감소시켜 ER proteostasis factor( 예: 샤페론 및 접는 효소)가 기존의 잘못 접힌 ER 단백질과 결합하고 ERAD 또는 autophagy를 포함한 메커니즘을 통해 재접힘 또는 제거를 촉진함으로써 ER을 보호합니다. (그림 3A ).
접힘 부하를 줄이는 것 외에도 PERK 의존성 번역 감쇠는 수명이 짧은 단백질의 분해 증가와 같은 메커니즘을 통해 세포 주기 진행, 미토콘드리아 단백질 수입 및 미토콘드리아 형태를 포함한 세포 생리학의 다른 측면을 조절하는 것으로 나타났습니다.
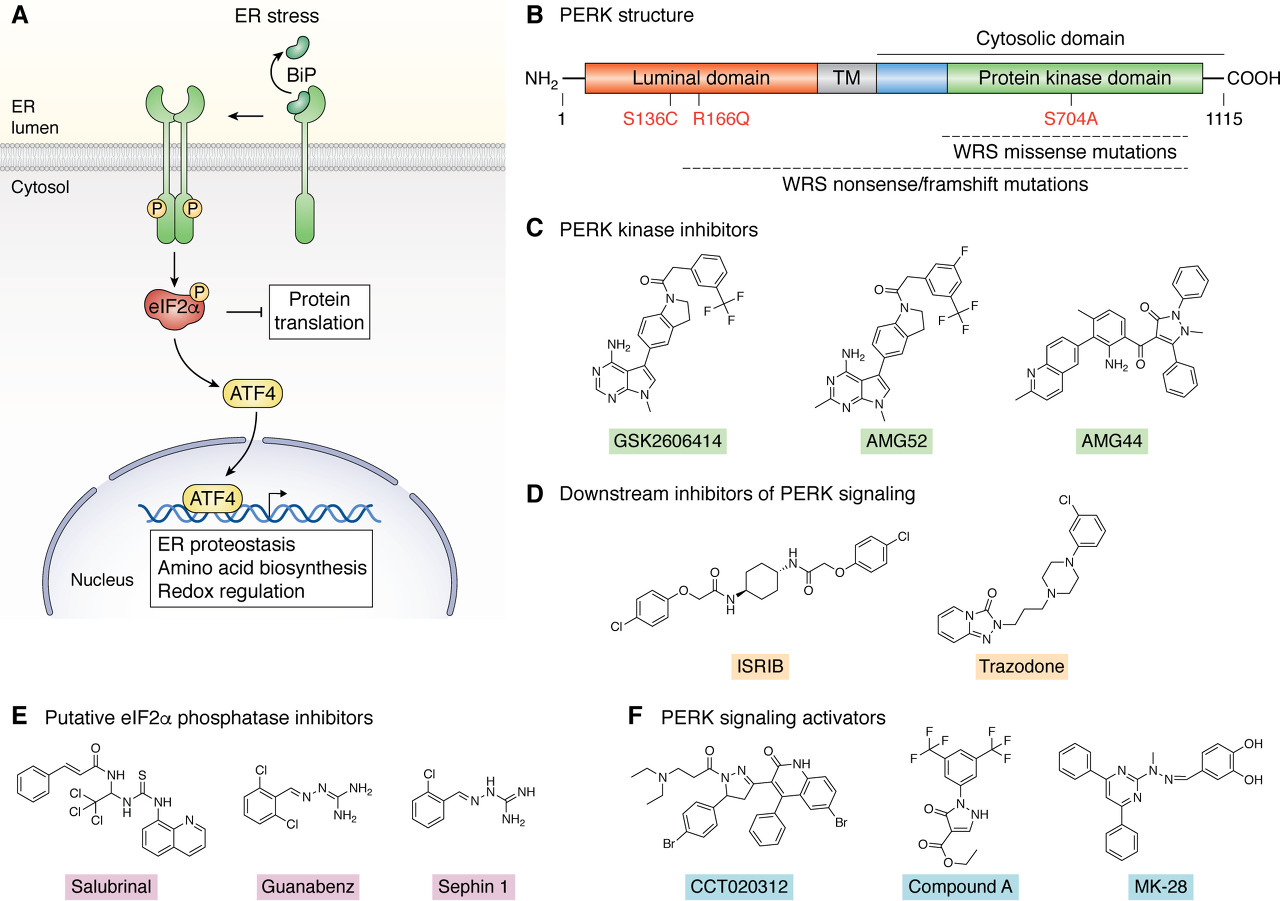
약리학적 PERK 조절 화합물.
A , UPR의 PERK 시그널링에 대한 활성화 및 시그널링 메커니즘.
B , 관강 도메인, 막횡단 도메인( TM ) 및 세포질 단백질 키나제 도메인을 포함하는 PERK의 도메인 구조 .
단백질 키나제 도메인 은 PERK 자가인산화와 인산화 eIF2α를 통해 A에 표시된 대로 기능하며 ,
후자는 PERK 신호전달 캐스케이드의 핵심 단계입니다.
C , PERK 키나제 억제제 GSK2606414, AMG52 및 AMG44의 구조. WRS , Wolcott-Rallison 증후군 관련 돌연변이가 표시됩니다.
D, eIF2α 인산화의 하류에서 PERK 신호 전달을 차단하는 PERK 신호 전달 ISRIB 및 트라조돈 억제제의 구조. E , 추정되는 eIF2α 포스파타제 억제제 살루브리날, 구아나벤즈 및 세핀 1의 구조 .
F , 표현형 및 컴퓨터 스크리닝 접근법을 통해 확인된 PERK 활성제 CCT020312, 화합물 A 및 MK-28의 구조.
일반적인 단백질 합성은 PERK 의존성 eIF2α 인산화에 의해 감소되는 반면, 이러한 조건에서는 특정 단백질 그룹이 선택적으로 번역됩니다. 여기에는 스트레스 반응성 전사 인자 활성화 전사 인자 4(ATF4)가 포함됩니다.그림 3가 )
ATF4는 세포 산화환원, 미토콘드리아 단백질 항상성, tRNA 전하 및 영양소 수송을 포함한 다양한 생물학적 기능에 관여하는 스트레스 반응 유전자의 발현을 유도합니다.그림 3가 )
ATF4는 또한 eIF2α의 탈인산화 및 PERK 신호 전달을 억제하는 음성 피드백 루프에서 번역 복원에 관여하는 eIF2α 포스파타제 조절기 소단위 성장 정지 및 DNA 손상 유도성 단백질 34(GADD34)의 발현을 유도합니다
이 전사 활동을 통해 PERK는 급성 손상에 대한 세포 적응 및 생존을 촉진합니다.
그러나 심각하거나 만성적인 ER 스트레스는 PERK의 다운스트림에서 pro-apoptotic 신호 전달을 촉진합니다
PERK가 세포 사멸을 촉진하는 한 가지 메커니즘은 전사 인자 C/EBP-동종 단백질 10(CHOP)의 상향 조절을 통한 것입니다
CHOP는 Bcl2-like protein 11(BIM) 및 DR5를 포함한 여러 pro-apoptotic 요인의 발현을 유도하여 고유한 세포자멸사 신호전달 캐스케이드 및 카스파제 활성화를 활성화합니다
PERK는 또한 번역 감쇠 후 단백질 합성의 회복과 관련된 산화 스트레스 증가, 세포 대사를 방해하는 microRNA의 발현 증가, 세포자멸사의 X-연관 억제제와 같은 항세포자멸 인자의 억제를 포함한 다른 메커니즘을 통해 세포 사멸을 촉진할 수 있습니다.
장기간의 ER 스트레스에 대한 응답으로 RIDD 활성이 감소하여 세포자멸사에서 이 "단절"을 해제하고 DR5 매개 세포자멸사 신호전달을 허용합니다.
이러한 유형의 통합은 다양한 수준의 ER 스트레스에 대한 응답으로 세포 운명을 조절하는 정교한 메커니즘을 제공합니다.
ER 스트레스에 대한 반응으로 적응과 생존을 모두 결정하는 PERK의 중요성을 고려할 때, PERK 활동의 증가 및 감소가 다양한 유형의 질병과 관련이 있다는 것은 놀라운 일이 아닙니다.
PERK의 과활성은 프리온병, 알츠하이머병 및 전두측두엽 치매를 포함한 많은 신경퇴행성 질환의 임상 샘플 및 마우스 모델에서 관찰됩니다
키나제 억제제를 사용하여 PERK 자가인산화 억제
PERK를 조절하는 초기 전략 중 하나는 활성화에 필요한 PERK 자가인산화 단계를 억제하기 위해 키나제 활성 부위를 표적화하는 데 중점을 둡니다.
PERK 키나제 억제제의 투여는 췌장 및 다발성 골수종 종양 이종이식 모델에서 종양 형성을 감소시켰고 프리온 질환 및 전측두엽 치매를 포함한 신경퇴행성 질환의 마우스 모델에서 결과를 개선했습니다.
그러나 이러한 화합물은 생체 내 에서 여전히 다소 제한적입니다.체중 감소 및 췌장 독성을 포함하여 용량 의존적 결함이 입증되었습니다.
그럼에도 불구하고, PERK 키나제 억제제는 질병 모델에서 PERK를 억제하는 치료 가능성을 정의하는 데 가치가 있는 것으로 입증되었으며 임상 사용을 위해 계속 탐색되고 있습니다.
PERK 신호 전달을 억제하기 위한 eIF2B의 약리학적 활성화
키나제 활성 부위를 직접 표적화하는 것과는 대조적으로, 다른 전략은 편향되지 않은 세포 기반 표현형 스크린을 사용하여 PERK 신호 전달을 억제하는 화합물을 식별합니다.
ISRIB와 유사한 방식으로 PERK 신호 전달 경로를 표적으로 하는 다른 화합물을 확인하는 데 상당한 관심이 있습니다. 한 가지 잠재적인 대안은 항우울제 선택적 세로토닌 흡수 억제제 트라조돈입니다(그림 3디 ). Trazodone은 eIF2α 인산화의 다운스트림에서 eIF2α 전사 및 번역 신호 전달을 차단하는 것으로 나타났습니다.
eIF2α 인산화 의존성 신호의 억제제로서 트라조돈의 잠재력을 완전히 이해하기 위해서는 추가 연구가 필요하지만( 예: 선택적 세로토닌 흡수 억제제 활성을 eIF2α의 신호 하류 신호에 미치는 영향으로부터 분리할 가능성), 트라조돈의 식별은 잠재력을 더욱 강조합니다.
질병에 개입하기 위한 유망한 전략으로 eIF2α 인산화의 다운스트림 PERK 신호를 표적화하기 위한 것입니다.
PERK-eIF2α 신호 전달을 향상시키기 위한 단백질 포스파타제 표적화
PERK 매개 eIF2α 신호 전달을 억제하는 것 외에도 이 경로를 통한 활성 강화는 인간 질병의 병리학적 결과를 개선할 수 있는 기회를 제공합니다.
PERK를 활성화하는 화합물을 개발하는 데 있어 중요한 과제는 이 경로에 의해 유도되는 세포자멸사 신호전달입니다. 세포자멸사 신호전달을 피하기 위해 PERK 활성을 증가시키는 전략은 주로 PERK 신호전달의 다운스트림 측면, 특히 eIF2α의 탈인산화를 담당하는 포스파타제를 표적으로 하는 데 초점을 맞추었습니다.
PERK 활성제
PERK 신호 전달을 선택적으로 활성화하는 소분자에 대한 초기 보고서는 G 1 /S 세포 주기 체크포인트 를 활성화하여 암세포 진행을 중단시키는 화합물을 식별하는 데 초점을 맞춘 인간 결장암 세포의 고처리량 스크리닝에서 비롯되었습니다
UPR의 ATF6
확인된 UPR의 마지막 부분은 ATF6 경로였습니다.
IRE1과 마찬가지로 인간은 두 개의 다른 ATF6 유전자인 ATF6 α와 ATF6 β를 암호화 합니다.
ATF6β는 주로 조절 역할을 하는 반면, ATF6α는 ER 스트레스에 반응하여 세포 생리를 적응시키는 일차 단백질입니다.
따라서 우리는 이 리뷰에서 주로 ATF6α에 대해 논의합니다. 여기에서는 ATF6이라고 부를 것입니다.
ATF6은 N-말단 bZIP 전사 인자 도메인과 C-말단 ER 내강 도메인을 포함하는 유형 II 막횡단 단백질입니다(그림 4, A 및 B ).
활성화를 위해 올리고머화 및 자가인산화에 의존하는 IRE1 및 PERK와 달리 ATF6은 다른 메커니즘을 통해 활성화됩니다.
ER 스트레스가 없는 경우 ATF6은 ER 내강에 국한된 단백질 이황화 이성화효소(PDI)에 의해 유지되는 단량체 및 이황화 결합 이량체/올리고머로 존재합니다.그림 4가 ).
산화된 ATF6은 ER HSP70 BiP에 의해 루미날 도메인에서 결합되고 ER 내에 유지됩니다(그림 4A ) .
ER 스트레스에 대한 응답으로 ATF6 이황화물은 PDI 의존성 메커니즘을 통해 환원되고 BiP는 내강 영역에서 방출되어 환원된 단량체 ATF6이 증가합니다.
이 감소된 ATF6 단량체는 골지로 이동되고 부위 1 및 부위 2 프로테아제(각각 S1P 및 S2P)에 의해 단백질 분해 처리됩니다.그림 4A ).
이것은 활성, N-말단 ATF6 bZIP 전사 인자 도메인을 방출하여 핵에 이합체화 및 국재화합니다. 이 활성 ATF6 전사 인자 는 표적 유전자 프로모터에서 ER 스트레스 반응 요소(ERSEs)를 결합 함으로써 다중 ER 단백성 인자( 예: BiP) 의 상향 조절을 포함하는 전사 반응을 유도합니다.그림 4A ).
ER proteostasis 외에도 ATF6 활성화는 각각 RHEB 및 카탈라아제를 포함한 전사 표적의 상향 조절을 통해 세포 성장 및 산화 환원 조절을 포함한 세포 생리학의 다른 측면을 전사적으로 조절합니다

ATF6 UPR 신호 전달 경로의 식별 및 표적화.
A , ATF6 활성화 및 다운스트림 전사 신호전달의 메커니즘.
B , N-말단 bZIP 전사 인자 도메인, 막횡단 도메인(TM) 및 내강 도메인을 포함하는 ATF6의 도메인 구조.
ATF6 활성화 시, 류신 지퍼 모티프를 함유하는 N-말단 세포질 도메인은 A에 도시된 바와 같이 골지체에서의 프로세싱을 통해 유리 되어 활성 ATF6 전사 인자의 방출을 초래한다.
achromatopsia와 관련된 ATF6의 특정 억제( 빨간색 ) 또는 활성화( 파란색 ) 돌연변이 가 표시됩니다.
C, S1P 억제제 PF429242 및 AEBSF 및 선택적 ATF6 억제제 Ceapin-A7을 포함한 ATF6 억제제의 구조.
D , ATF6 활성화 화합물 BiX, AA147 및 AA263의 구조.
ATF6 활성을 감소시키는 것이 스트레스가 없을 때 유기체 생리학에 크게 영향을 미치지 않는다는 것을 나타냅니다.
유사하게, 다양한 마우스 조직에서 활성 ATF6 전사 인자 도메인의 과발현은 잘 견디며 조직 특이적 독성과 관련이 없습니다.
대신, 증가된 ATF6 전사 활성은 당뇨병, 단백질 접힘 질환, 심근경색, 뇌졸중을 비롯한 여러 질병의 세포 및 설치류 모델에서 보호적인 것으로 나타났습니다
약리학적 UPR 조절제로부터 배운 교훈
특정 UPR 신호 전달 경로에 대한 화합물 선택성
암 선택적 UPR 활성화제 또는 억제제를 설정하는 데 있어 가장 중요한 문제 중 하나는 특정 UPR 경로에 대한 선택성을 정의하는 것입니다.
표적을 벗어난 활성과 관련된 독성은 위에서 논의한 바와 같이 특정 UPR 조절 화합물의 적용을 제한합니다. 따라서 개별 UPR 신호 전달 경로를 표적으로 하는 새로운 화합물을 식별하고 개발할 때 화합물 선택성과 그 한계를 이해하는 것이 중요합니다.
UPR 응답을 "재형성"하는 부분 변조기
UPR 조절제 개발의 또 다른 주목할만한 과제는 특정 계통을 통한 신호 전달이 너무 많거나 너무 적으면 병리학으로 이어질 수 있다는 사실과 관련이 있습니다.
따라서 UPR 경로를 완전히 억제하거나 활성화하는 소분자는 둘 다 독성을 유발할 수 있습니다.
이 표적 독성은 암과 같은 특정 질병의 맥락에서 유익할 수 있지만 심각한 부작용( 예: PERK 키나제 억제제와 관련된 췌장 독성) 으로 인해 다른 질병에 대한 UPR 조절 화합물의 개발을 막을 수 있습니다.
이 문제에 대한 한 가지 해결책은 부분적으로 만 UPR 변조기의 개발에 있습니다.
UPR의 특정 부분을 통해 신호를 변경합니다.
이러한 유형의 화합물은 주어진 경로의 완전한 활성화 또는 억제와 관련된 독성을 종종 피할 수 있는 방식으로 UPR 반응을 "재형성"할 수 있는 독특한 기회를 제공합니다.
UPR 조절 화합물의 표현형 선택성
UPR 조절 화합물을 세포 및 생체 내 모델에 적용할 때 또 다른 중요한 고려 사항 은 표현형을 특정 UPR 경로의 활성화 또는 억제로 귀인하는 것과 관련된 문제입니다.
***********************************************************************
2020
건강과 질병에서 면역과 내성의 균형을 맞추는 데 있어 소포체 스트레스의 새로운 역할: 메커니즘 및 기회
https://www.frontiersin.org/articles/10.3389/fimmu.2019.03154/full
The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Op
The endoplasmic reticulum (ER) is an organelle equipped with mechanisms for proper protein folding, trafficking, and degradation to maintain protein homeostasis in the secretory pathway. As a defense mechanism, perturbation of ER proteostasis by ER stress
www.frontiersin.org
소포체(ER)는 분비 경로에서 단백질 항상성을 유지하기 위해 적절한 단백질 폴딩, 트래피킹 및 분해를 위한 메커니즘을 갖춘 소기관입니다.
방어 기작으로서, ER 스트레스 인자에 의한 ER 단백형성의 섭동은 ER에서 UPR(unfolded protein response)로 알려진 핵으로의 신호 전달 경로의 캐스케이드를 활성화합니다.
UPR의 주요 목표는 세포 생존을 위한 ER 항상성을 회복하기 위해 전사 및 번역 프로그램을 유도하는 것입니다.
따라서 UPR 신호 전달의 결함은 대사 질환, 퇴행성 질환, 염증성 질환 및 암을 비롯한 여러 질병의 주요 원인으로 연루되어 있습니다.
점점 더 많은 증거가 운명과 면역 반응의 크기를 조절하는 ER 스트레스의 중요한 역할을 뒷받침합니다.
게다가, 여러 UPR 약리학적 억제제의 이용 가능성은 UPR을 표적으로 하는 것이 질병의 면역 조절 및 면역 요법을 위한 새로운 전략이 될 수 있다는 희망을 높입니다.
서론: ER 스트레스, 면역 및 질병
소포체(ER)는 모든 진핵 세포의 분비 경로에서 단백질 품질 관리에 참여하는 필수 소기관입니다( 1 , 2 ). ER 항상성은 단백질 접힘, 칼슘 항상성, 지질 대사, 세포 분화 및 단백질 전위를 포함한 다양한 세포 내 생리 기능을 제어하는 데 중요합니다( 3 , 4 ).
그러나 영양 결핍, 저산소증, 산-염기 불균형, 활성 산소 종(ROS) 축적과 같은 특정 상황에서는 ER 기능의 용량이 초과되어 잘못 접힌 단백질이 축적될 수 있습니다..
그런 다음, 스트레스와 기능 장애를 해결하기 위해 ER의 진화적으로 보존되고 보호되는 메커니즘 중 하나인 펼쳐진 단백질 반응(UPR)이 뒤따릅니다.
분자적으로 UPR은 여러 세포 내 반응을 시작할 수 있습니다.
먼저, 단백질 처리 및 재접힘을 촉진하기 위해 단백질 샤페론의 상향 조절을 선호하면서 전반적인 단백질 합성이 약화됩니다.
증가된 샤페론이 접힘/재접힘 요구를 충족할 수 없는 경우 UPR은 ER 관련 분해 경로를 통해 단백질 이화작용을 유발합니다.
UPR 메커니즘은 또한 지질 대사를 촉진하여 ER 막의 합성을 증가시켜 ER의 물리적 공간을 확장하는 방향으로 조정됩니다.
마지막으로, 이러한 메커니즘이 만성 ER 스트레스를 역전시키지 못하면 UPR은 세포 사멸을 통해 세포 사멸을 유도합니다.
자가 면역 및 암과 같은 질병은 불균형한 면역 반응의 결과로 간주됩니다.
면역 세포 항상성을 유지하는 것이 이펙터와 조절 기능의 균형을 유지하는 데 중요하다는 점을 감안할 때, 새로운 증거는 ER 스트레스, 특히 면역 구획에서 신경 퇴행, 염증, 대사 장애 및 전염병을 포함한 다양한 병리학에 참여함을 시사합니다.
또한 ER 스트레스는 2형 대식세포(M2), 골수 유래 억제 세포(MDSC), 관용성 수지상 세포(DC) 등을 포함한 면역억제 세포의 조절을 통해 항종양 면역을 방해합니다.
세포 항상성과 질병에서 주요 UPR 센서의 역할
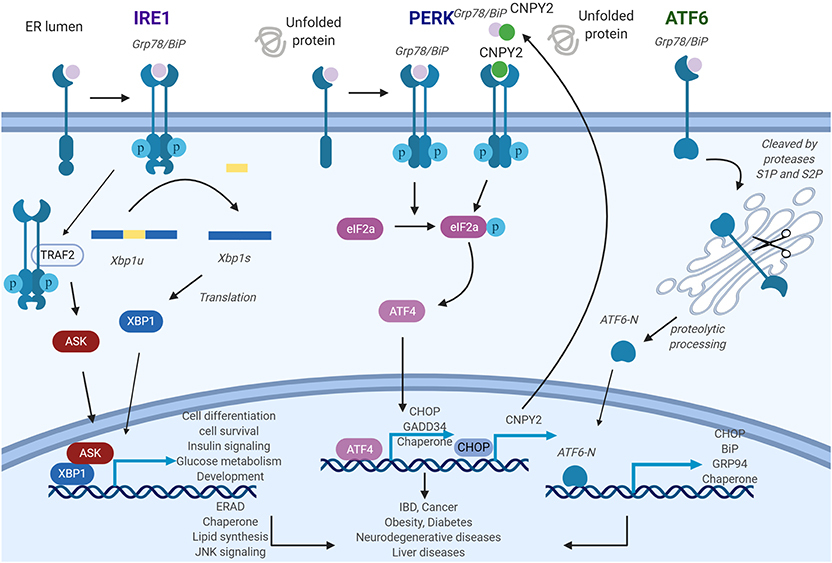
그림 1. UPR 경로에서 소포체(ER) 스트레스 센서의 일반적인 역할
이노시톨-요구 효소 1(IRE1),
PKR 유사 ER 키나제(PERK) 및
활성화 전사 인자 6(ATF6)은 ER 내강에서 세포질로의 ER 스트레스 신호를 전달합니다
IRE1 경로: ER 스트레스는 IRE1 올리고머화 및 자가인산화를 유도한 다음 활성화된 IRE1에 의해 XBP1의 스플라이싱이 촉발됩니다.
전사 인자로서 X-box 결합 단백질 1(XBP1)은 UPR 관련 유전자를 활성화합니다.
PERK 경로: 활성화된 PERK는 eIF2a를 인산화하고 ATF4를 추가로 자극하여 표적 유전자 발현을 조절합니다. 캐노피 동족체 2(CNPY2)는 Grp78에서 분리된 다음 PERK 오토키나제 활성을 촉진할 수 있습니다.
CAAT/인핸서 결합 단백질 상동 단백질(CHOP)의 번역 증가는 CNPY2 프로모터를 활성화하고 CNPY2 발현을 추가로 높입니다.
ATF6 경로: ATF6은 프로테아제 S1P 및 S2P에 의해 절단되어 ATF6-N을 생성합니다.
그런 다음 ATF6-N은 핵으로 이동하여 표적 유전자의 전사를 시작합니다.
IRE1-XBP1, PERK 및 ATF6 경로는 장기간 지속되면 다양한 질병의 발병에 기여할 수 있습니다.
IRE1
ER 스트레스 신호는 ER 스트레스 센서로 알려진 ER 막의 3가지 보존된 막횡단 단백질에 의해 크게 변환됩니다: 이노시톨-요구 효소 1(IRE1), PKR 유사 ER 키나제(PERK) 및 전사 인자 6 활성화(ATF6).
(IRE1)은 IRE1α 및 IRE1β의 두 가지 동형으로 구성됩니다.
IRE1α 유전자 녹아웃 마우스는 배아적으로 치명적이지만 IRE1β 녹아웃은 비중복 기능적 역할을 나타내는 심각한 표현형 이상을 나타내지 않습니다.
실제로, IRE1α가 어디에나 존재하는 반면 IRE1β 발현은 폐 점막 상피와 위장관으로 제한되기 때문에 이소형의 발현 패턴이 이를 입증하는 것으로 보입니다.
IRE1은 펩타이드 인식 및 세린/트레오닌 키나제 활성을 위한 ER-루미날 센서가 있는 유형 I 막횡단 단백질입니다( 그림 1 ).
펼쳐진 단백질(unfolded proteins)의 축적은 IRE1의 내강 도메인에서 면역글로불린 결합 단백질(BiP)의 해리를 유발하여 즉각적인 IRE1 올리고머화 및 키나제 도메인의 자가인산화를 유발합니다( 21 , 22 ).
이러한 구조적 변화에 대한 반응으로 IRE1 RNase 도메인이 활성화되어 X-box 결합 단백질 1 ( XBP1) 의 비정상적인 세포질 접합(잘라이음)을 유도합니다.
ER 항상성을 회복하기 위해 XBP1s는 ER 관련 분해 경로에서 단백질 폴딩 샤페론과 이펙터 분자를 포함한 다양한 표적 유전자의 전사를 자극합니다.
항상성을 유지하는 것 외에도 XBP1은 세포 분화, 생존, 인슐린 신호 전달, 포도당 대사 및 발달과 같은 여러 세포 신호 전달 경로에도 참여합니다.
최근에 RNase 활성의 활성화는 XBP1의 비 전통적인 접합을 증가시킬 뿐만 아니라그러나 조절된 IRE1 의존성 붕괴(RIDD)라고 하는 별개의 메커니즘을 통해 여러 다른 전사체를 표적으로 삼기도 합니다.
RIDD는 단백질 접힘 및 ER 스트레스 조절에 관여하는 단백질을 암호화하는 mRNA를 선택적으로 절단하고 RIDD 신호전달의 만성 활성화는 세포 사멸 메커니즘을 촉진합니다.
엔도뉴클레아제 활성 외에도 IRE1은 IRE1과 (TNF) 및 (TRAF2)의 직접적인 상호작용을 통해 JNK 신호 전달을 활성화합니다.
이 IRE1-TRAF2 복합체는 세포 사멸 신호 조절 키나제 1(ASK1)을 모집하고 활성화하여 궁극적으로 세포 사멸을 유발하는 (JNK) 경로를 활성화합니다.
수많은 연구에서 면역과 염증에서 ER 스트레스 반응의 중요성이 밝혀졌습니다. 가장 잘 연구된 ER 스트레스 관련 염증성 질환 중 하나는 염증성 장 질환(IBD)입니다.
IBD는 뚜렷한 임상 징후와 병리학을 갖지만 근본적인 병인을 복잡하게 하는 장의 인간 만성 염증성 장애입니다.
XBP1은 또한 대식세포 및 DC와 같은 다양한 세포 유형에서 염증에 역할을 합니다.
ER 스트레스는 인터루킨-1(IL-1), IL-6, IL-8, TNFα 및 단핵구 화학 유인 단백질 1(MCP1)을 포함한 사이토카인 생성을 증가시킵니다.
만성 염증 상태인 종양 미세 환경(TME)은 높은 수준의 ER 스트레스가 특징입니다.
암 세포 기능을 본질적으로 조절하는 것 외에도 IRE1은 TME의 면역 세포를 심도 있게 조절합니다.
염증 조절 외에도 IRE1 경로는 비만 및 당뇨병을 포함한 대사 질환과도 관련이 있습니다( 42 ).
고지방식이(HFD) 유도 비만 마우스 모델 및 렙틴 결핍 마우스 모델과 같이 잘 정립된 여러 마우스 비만 모델을 사용하여 비만은 지방 조직 및 간에서 인산화된 IRE1, PERK 및 JNK의 발현 증가와 관련이 있음을 발견했습니다. .
XBP1 결핍 마우스는 WT 대조군과 비교할 때 손상된 포도당 항상성을 나타냅니다.
XBP1s는 JNK의 과활성화와 인슐린 수용체 하위 상태-1(IRS-1)의 인산화를 통해 인슐린 수용체 신호 전달을 억제합니다.
췌장 β 세포 특이적 XBP1 결핍 모델은 감소된 인슐린 분비로 인한 포도당 불내성을 나타내며, 이는 대사 질환에서 ER 스트레스의 중요한 역할을 암시합니다.
PERK
PERK는 C-말단에 세린/트레오닌 키나제 활성이 있는 유형 1 막횡단 단백질이며 관강 도메인 센서에 의해 잘못 접힌 단백질의 축적을 인식합니다.
PERK의 활성화는 내강 도메인에서 BiP의 해리에 의해 시작되어 올리고머화 및 자가인산화가 발생합니다.
활성 PERK는 eIF2α를 인산화하여 일반적인 단백질 합성을 감소시켜 ER로 들어가는 단백질의 부하를 줄입니다.
이 빠른 반응은 생존 메커니즘으로 작용합니다.
놀랍게도, 이러한 상황에서 ATF4(전사 인자 4 활성화)와 같은 일부 전사체는 더 효율적으로 번역됩니다.
PERK는 처음에는 생존 메커니즘을 활성화하지만 CHOP 및 GADD34를 조절하여 장기간의 ER 스트레스 하에서 proapoptotic 메커니즘으로 전환합니다.
우리의 발견은 특히 PERK 분기에 대한 UPR 개시의 새로운 메커니즘을 밝혀냈습니다.
PERK 활성화는 BiP 해리와 CNPY2에 대한 직접 결합 모두에 의해 촉발될 수 있습니다.
당연히 CNPY2 결실은 비알코올성 지방간 질환(NAFLD)으로부터 마우스를 보호합니다.
염증 및 면역 반응에서 CNPY2의 역할은 활발한 조사를 받고 있습니다.
PERK 경로는 특히 신경퇴행성 질환 및 대사성 질환에서 다양한 질병에 연루되어 있습니다.
ER 스트레스는 타우 단백질의 분해를 지연시키고 타우의 과인산화를 일으켜 UPR을 더욱 증폭시켜 악순환을 만듭니다.
신경퇴행성 질환의 또 다른 주요 단백질은 아밀로이드 β입니다.
아밀로이드 β 올리고머 또는 섬유소는 신경 세포에서 PERK 경로를 촉발하고 추가 조사에 따르면 Ca 2+ 가 이 작용의 가능한 매개체로 밝혀졌습니다.
대사 질환에서 PERK는 eIF2a를 인산화하여 ATF4 전위를 유도하고 세포 번역을 억제합니다.
PERK 경로는 또한 지방세포에서 IKKβ 경로를 조작하고 염증성 사이토카인 생성을 촉진합니다.
또한, 마우스 뇌 성상교세포에서 PERK의 활성화는 IL-6 발현 증가를 통해 뇌 염증을 가속화하는 것으로 나타났으며 이 과정은 JAK/STAT3 신호 전달 경로에 의해 조절되었습니다.
PERK는 면역 세포를 조절하는 데 중요한 역할을 하며 이는 나중에 논의될 주제입니다.
ATF6
활성화 전사 인자 6 ( ATF6)은 ATF6α 및 ATF6β로 구성된 류신 지퍼 전사 인자 및 II형 ER 막관통 단백질( leucine zipper transcription factor and a type II ER transmembrane protein )입니다.
흥미롭게도 ATF6의 α 또는 β 소단위의 유전적 결실은 생존력에 영향을 미치지 않으며, 이는 단백질 소단위의 중복 또는 보상 역할을 시사합니다
ER에 잘못 접힌 단백질이 축적되면 BiP가 ATF6에서 해리되어 잘못 접힌 단백질과 BiP의 상호 작용이 가능합니다
그 후, 유리 ATF6은 ER에서 골지로 이동하여 두 개의 다른 프로테아제에 의해 매개되는 절단을 겪습니다.
그러면 ATF6의 절단된 N-말단 세포질 도메인이 핵으로 이동하여 BiP, Grp94 및 CHOP를 포함한 표적 유전자를 활성화하여 ER에서 개선된 단백질 접힘 활성을 유도합니다.
또한 ATF6은 마이크로 RNA 발현 수준을 조절하는 것으로 나타났습니다.
높은 ATF6 발현은 마우스 모델에서의 작업과 일치하는 결장직장암 환자의 불량한 예후와 상관관계가 있습니다.
활성화된 ATF6의 상피 세포 특이적 과발현이 있는 마우스 모델은 자발적으로 결장 선종을 발생시켰습니다.
ATF6은 또한 생체 내에서 mTOR 신호 전달을 조절하여 화학 요법 내성에 기여합니다 .
다른 ER 스트레스 요인과 유사하게 ATF6 null 마우스는 고지혈증과 인슐린 저항성이 발생하기 쉽습니다.
ATF6 녹아웃 마우스는 또한 간 기능 장애와 지방증을 나타냅니다
ER 스트레스의 면역학적 영향
ER 스트레스는 단백질 접힘, 칼슘 항상성, 지질 대사, 세포 분화 및 단백질 전위를 포함한 다양한 세포 내 생리 기능을 제어하는 데 중요한 역할을 합니다.
따라서 ER 스트레스 반응의 오작동은 당연하게도 선천성 및 후천성 면역 반응의 조절 장애와 관련이 있습니다.
최근 연구에 따르면 UPR 센서는 T 세포, B 세포, DC, 대식세포 및 MDSC를 포함한 여러 면역 세포 유형의 발달, 분화, 활성화, 사이토카인 생산 및 세포자멸사를 조절하는 데 관여합니다( 그림 2 , 3).
따라서 면역 구획에서 UPR 이펙터의 새로운 역할은 암을 비롯한 여러 면역 장애의 관리에서 UPR을 표적화할 가능성을 높입니다.
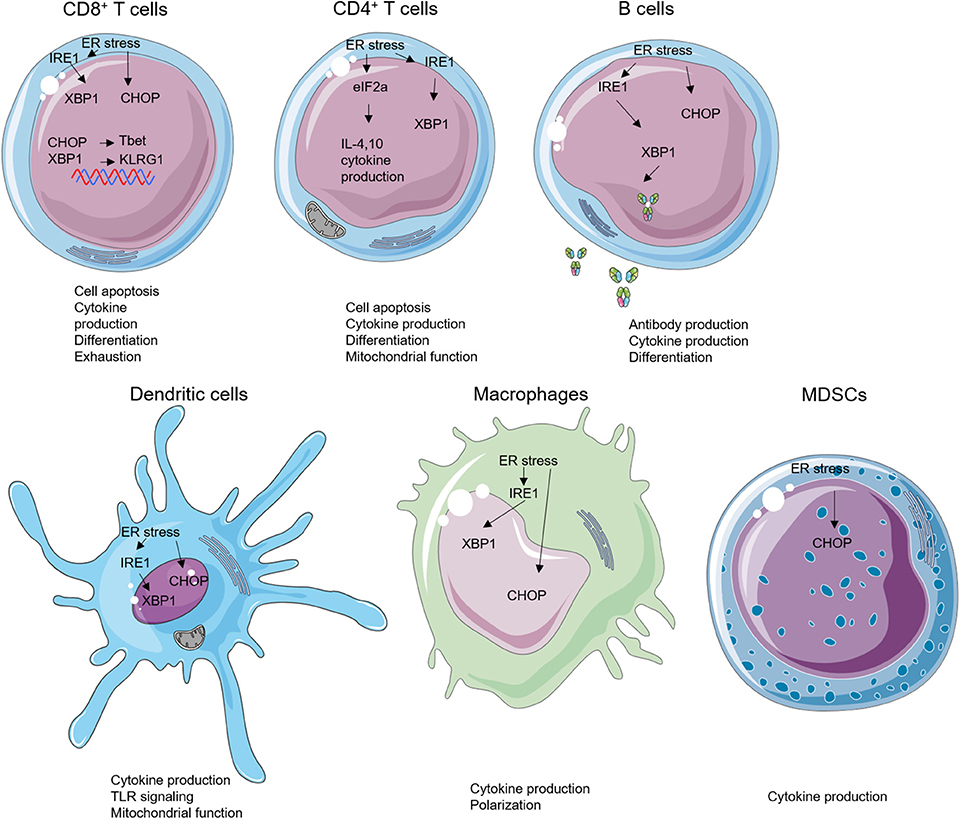
그림 2 . 면역 세포의 소포체(ER) 스트레스 및 펼쳐진 단백질 반응(UPR) 이펙터.
ER 스트레스는 세포 사멸, 사이토카인 생산, 세포 분화, 항체 생산, 미토콘드리아 기능 및 톨 유사 수용체(TLR) 신호와 같은 면역 세포의 다양한 하위 집합의 생물학을 조절할 수 있습니다.

그림 3 . 소포체(ER) 스트레스는 염증에서 다면적인 역할을 합니다.
ER 스트레스는 주요 면역 세포를 조절하여 염증 촉진 및 항염의 항상성 환경을 설정합니다.
CD8 + T 세포
형질전환된 종양 세포 또는 세포내 병원체에 감염된 세포는 T-세포 수용체 결합 및 후속적인 활성화 및 사이토카인 분비를 통해 세포독성 CD8 + T 세포에 의해 인식되고 제거될 수 있다 .
ER 스트레스는 CD8 + T 세포 의 분화, 사이토카인 생산, 고갈 및 세포자멸사를 조절 합니다.
급성 바이러스 또는 박테리아 감염 후 항원 특이적 CD8 + T 세포 집단의 확장이 일어나고 이 과정은 접합된 XBP1 mRNA의 수준을 증가시킵니다.
뮤린 B16 흑색종 TME에서 높은 수준의 콜레스테롤은 지질 대사를 방해하고 CD8 + T 세포 에서 XBP1의 발현을 자극 합니다.
또한 XBP1을 억제하거나 콜레스테롤을 감소시키면 생체 내에서 CD8 + T 세포 의 항종양 효과가 향상됩니다.
또한 Cao et al. CHOP는 CD8 + T 세포 의 이펙터 및 미토콘드리아 기능을 부정적으로 조절한다는 것을 입증했습니다 .
따라서 CD8 + 에서 CHOP의 삭제T 세포는 T-bet 전사의 직접적인 증가를 통해 항종양 면역 반응을 향상시킵니다.
ER 스트레스는 여러 질병 환경에서 CD8 + T 세포 대사 조절 및 활력과 관련이 있습니다.
실제로 미토콘드리아 기능은 CD8 + T 세포에 필수적입니다.
CD8 + T 세포 에서 PERK의 유전적 및 약리학적 억제는 프로그램된 세포 사멸 단백질-1+ CD8 + 종양 침윤 림프구(TIL) 에서 미토콘드리아 ROS 생성 을 중단하여 CD8 + 종양 침윤 림프구 생존력을 강화하고 항종양 면역을 강화합니다.
CHOP, 인산화된 eIF2α 및 GRP78과 같은 여러 ER 스트레스 센서는 T 세포 사멸을 유도하여 ER 스트레스가 세포 사멸을 차단하고 T 세포 지속성을 증가시킬 수 있음을 암시합니다.
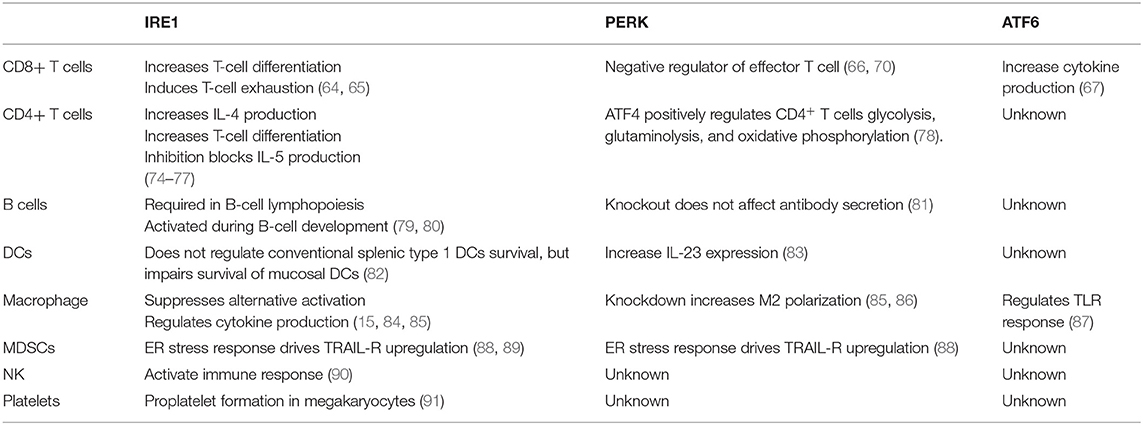
표 1 . 특정 면역 세포 집단에서 펼쳐진 단백질 반응(UPR) 이펙터의 역할.
CD4 + T 세포
정상적인 CD4 + T 세포는 다양한 조건에서 Th1, Th2, Th17 및 조절 T 세포(Treg)를 비롯한 T 헬퍼(Th) 세포의 다양한 하위 집합으로 분화할 수 있습니다( 92 , 93 ).
ER 스트레스는 CD4 + T 세포 의 분화, 가소성, 효과기 기능 및 세포자멸사를 조절하는 데 중요한 역할을 합니다 .
eIF2α의 인산화를 통한 ER 스트레스의 활성화는 Th2 활성화 및 분화 과정에서 발생하며, 차례로 인산화된 eIF2α의 축적은 Th2 프라이밍 동안 IL-4의 방출을 향상시킵니다
XBP1s 활성은 Th17 분화를 촉진하고 생쥐에서 실험적 자가면역 뇌척수염(EAE)을 촉진하는 데 중요한 기능을 합니다.
XBP1의 유전적 및 약리학적 억제는 Th17에서 인터페론 감마(IFNγ) 생성을 감소시켜 EAE의 발병을 감소시킵니다
PERK 경로 조절자 ATF4는 CD4 + T 세포 해당 작용을 강화 하고 mTORC1 활성화를 조절합니다.
ATF4 결핍 CD4 +T 세포는 EAE 모델에서 Th1 대신 Th17 세포로 분화되는 경향이 있습니다.
우리 그룹은 ER 스트레스가 CD4 + T 세포 활성화와 그 기능에 중요하다는 것을 보여주었습니다 .
CD4 + 의 IRE1α-XBP1 경로T 세포는 또한 난소암의 항종양 면역과 관련이 있습니다.
이펙터 T 세포 분화 및 기능에서 UPR의 역할 외에도 ER 스트레스는 또한 T 세포 자가포식 및 세포자멸사를 조절합니다.
또한, ER 스트레스 유발 ROS는 인간 T 세포의 세포 사멸 및 기능 장애를 촉진합니다
이 모든 역할 외에도 CD4 +T 세포는 또한 신경 염증을 유발하는 데 도움이 될 수 있습니다.
뇌로의 CD4 + T 세포 침윤은 다발성 경화증 및 파킨슨병을 비롯한 특정 경우에 염증을 증가시킬 수 있습니다.
그러나 CD4 + T 세포는 감염, 뇌졸중 및 신경퇴행성 질환에 대한 신경보호 역할도 할 수 있습니다.
ER 스트레스가 있는 뇌 염증에서 CD4 + T 세포 의 이러한 이중 역할은 추가 조사가 필요합니다
B 세포
UPR 이펙터는 형질 세포로 분화하는 동안 B 세포에서 상승하며 효율적인 항체 생산에 필요합니다
특히, XBP1은 형질 세포에서 UPR을 유도하고 면역글로불린 합성을 촉진합니다.
XBP1이 결핍된 B 세포는 정상적으로 발달하고 활성화될 수 있지만 면역글로불린을 생성하지 못합니다.
이러한 발견은 XBP1과 그 하류 분자가 B 세포 분화와 면역 글로불린 생산을 조절한다는 것을 나타냅니다.
XBP1과 달리 IRE1α는 B 세포에서 분화와 항체 생산을 모두 조절합니다.
IRE1α는 또한 Ig 유전자 재배열 및 B 세포 수용체 생성에 필요한 B 세포 림프구 형성에서 중요한 역할을 합니다.
IRE1α 결핍 B 세포는 프로 B 세포 단계를 넘어 발달할 수 없었습니다
또한, ER 스트레스는 B 세포를 활성화하여 전염증성 사이토카인의 생산을 조절합니다.
이러한 관찰은 정상 조건 및 질병에서 B 세포 분화 및 기능에서 ER 스트레스의 잠재적 역할을 나타냅니다( 표 1 ).
수지상 세포 (Dendritic Cell)
전문 항원 제시 세포 역할을 하는 DC는 적응 면역 반응의 시작에 중요하며 DC는 ER 스트레스에 의해 엄격하게 조절됩니다.
XBP1의 발현은 TLR(Toll-like receptor) 신호에 대한 반응으로 DC의 성숙 동안 증가합니다
XBP1은 또한 과량의 지방산이 주변 환경에 축적되어 해당 작용 장애를 초래하는 상황에서 미토콘드리아 ROS를 통해 IL-23 생성을 촉진할 수 있습니다
TME에서 침윤성 DC는 심한 ER 스트레스를 받아 항종양 면역을 손상시킵니다.
DC에서 XBP1을 표적으로 하면 여러 모델에서 항종양 면역이 향상됩니다.
Glimcher 그룹은 종양 관련 DC(tDC)에서 XBP1의 DC 특이적 결실 또는 선택적 나노입자 매개 XBP1 침묵이 T 세포 매개 항종양 면역을 증가시킨다는 것을 입증했습니다.
이 연구는 또한 XBP1이 tDC에서 트리글리세리드 생합성 프로그램을 상승시켜 비정상적인 지질 축적과 항원 제시 감소를 초래한다는 것을 보여주었습니다.
DC에서 XBP1을 유전적으로 및 약리학적으로 표적화하면 관용성 DC를 면역원성 DC로 전환합니다
IRE1α 엔도뉴클레아제 기능의 약리학적 억제는 주요 조직적합성 복합체 클래스 II에 대한 종양 항원의 제시를 손상시키지 않으면서 주요 조직적합성 복합체 클래스 I 경로에 대한 종양 관련 항원의 교차 제시를 선택적으로 차단하여 CD8 + T-세포 프라이밍 을 억제합니다
이러한 대조적인 관찰은 DC의 ER 스트레스를 질병과 연결하는 데 있어 잠재적인 함정에 주의를 기울입니다( 표 1 ).
대식세포 (Macrophages)
대식세포는 병원체를 포식하고 염증성 사이토카인을 생성하는 기능을 하는 타고난 면역에 관여하는 중요한 세포 유형입니다.
대식세포의 분극화는 염증성 또는 항염증성 사이토카인을 생성하는 기능에 중요합니다.
IRE1α의 특정 절제는 대사 질환에서 M1-M2 불균형을 유도합니다.
또한 유도된 XBP1 발현은 대식세포에서 전염증성 사이토카인(IL-6, TNFα, IFNβ) 생성을 촉진합니다
XBP1과 달리 ATF6은 TLR 신호전달 조절을 통해 대식세포에서 염증성 사이토카인 생성을 조절하지만 ATF6은 일반적으로 TLR 신호전달에 영향을 받지 않습니다
더욱이, 대식세포에서 ATF6의 녹다운은 핵인자 카파 B의 활성을 제한함으로써 특히 TNFα 및 IL-6 발현을 감소시켰습니다.
전반적으로, TME에서 M2 대식세포를 재분극시키는 수단으로 ER 스트레스를 표적으로 하는 것은 매력적인 치료 접근법일 수 있습니다.
MDSC
MDSC는 이질적인 표현형과 기능을 가진 TME의 면역 억제 세포 그룹입니다.
다른 골수 집단과 마찬가지로 ER 스트레스는 암에서 MDSC의 기능을 조절하는 데 중요한 역할을 합니다.
저용량 Tg의 다중 투여는 ER 스트레스를 유발하고 아르기나제-1, 유도성 산화질소 합성효소 및 NADPH 산화효소 2 생성을 상향 조절함으로써 종양 침윤성 MDSC의 면역억제 능력을 향상시켜 종양에 상주하는 CD8 + T 세포 세포독성을 전반적으로 손상시킬 수 있습니다.
기타 면역 세포
자연 살해(NK) 세포는 숙주 면역학적 항상성과 병인의 중요한 조절자입니다.
Glimcher 그룹은 IRE1-XBP1 경로가 c-Myc의 직접적인 조절을 통해 NK 세포 증식을 매개한다는 것을 보여주었습니다.
또한 XBP1은 NK 세포의 산화적 인산화를 촉진합니다
우리 그룹은 혈소판이 항종양 면역에서 중요한 매개체임을 보여주었습니다
혈소판의 형성은 ER 스트레스에 의해 매개됩니다.
ER 스트레스 요인을 사용하여 거핵구에서 caspase-4가 억제되어 혈소판 생성이 향상되었습니다
그러나 자세한 메커니즘은 아직 명확하지 않습니다. 전반적으로 NK 세포와 혈소판에서 ER 스트레스의 역할은 아직 파악되지 않았습니다( 표 1 ).
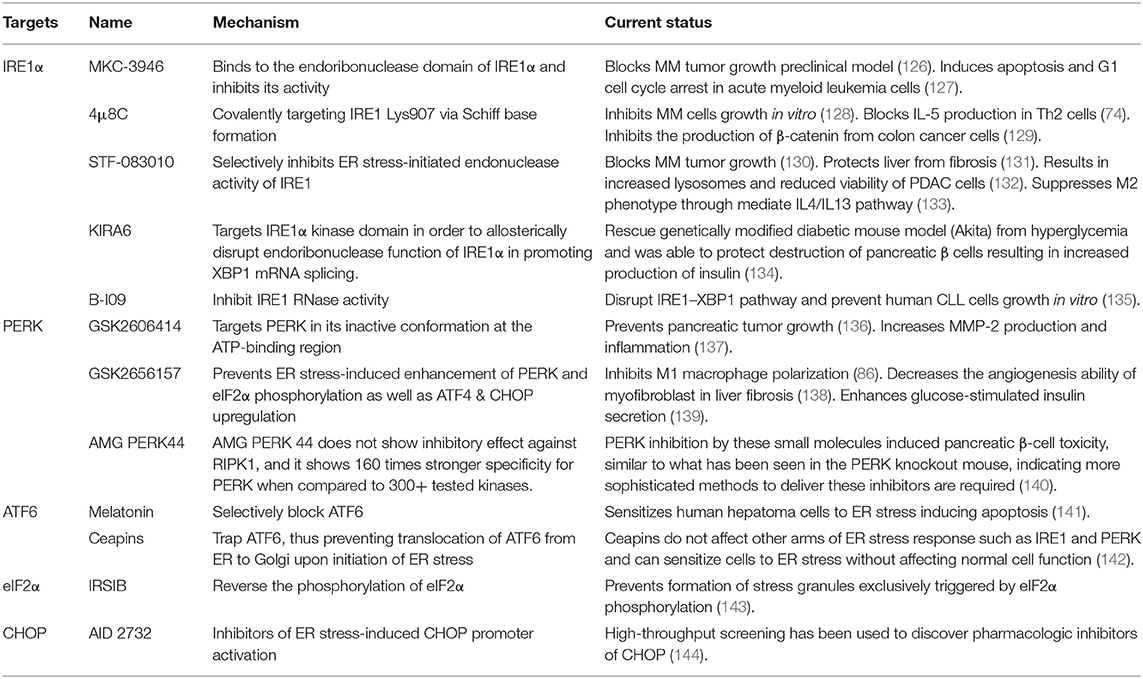
표 2(UPR) 억제제의 전임상 사용.
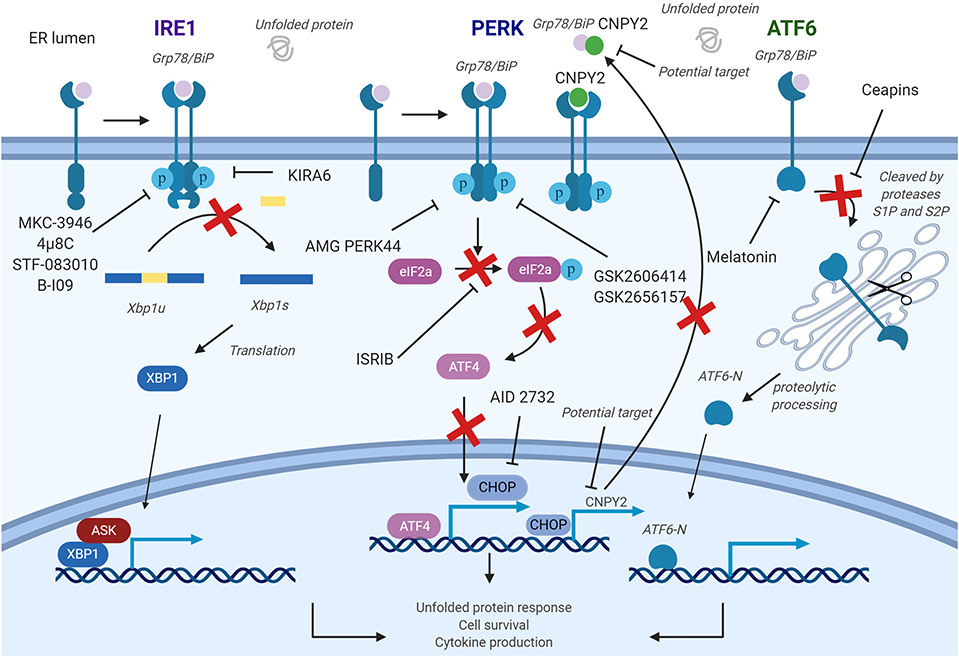
그림 4. 질병에서 소포체(ER) 스트레스를 조절하기 위한 약리학적 전략.
(IRE1a) 키나제 도메인은 4μ8c, MKC-3946 및 B-I09와 같은 소분자 약물에 의해 직접 억제될 수 있습니다.
이러한 화합물은 Xbp1 메신저 RNA(mRNA)의 접합을 방지합니다.
IRE1의 ER 스트레스 개시 엔도뉴클레아제 활성을 선택적으로 억제하는 화합물인 STF-08310은 또한 추가 다운스트림 신호 전달을 방지합니다.
이러한 화합물은 T 세포, B 세포, 수지상 세포(DC), 대식세포, 골수 유래 억제 세포(MDSC) 및 자연 살해(NK) 세포에 영향을 미칠 수 있습니다.
(PERK) 억제제는 PERK 및 그 다운스트림 인자의 향상을 억제하기 위해 개발되었으며 T 세포, B 세포, DC 및 대식세포를 표적화하는 데 사용될 수 있습니다.
게다가, CHOP 특이적 억제제는 CHOP 프로모터 활성화를 방지하기 위해 개발되었습니다.
멜라토닌은 또한 ATF6을 선택적으로 억제하는 것으로 보고되었습니다.
PERK-CHOP 경로는 CNPY2(canopy homolog 2) 억제에 의해 차단될 수도 있습니다.
UPR은 ER 항상성을 위한 필수 체크포인트이며 ER에서 잘못 접힌 단백질의 축적으로 인한 스트레스에 대한 생리학적 센서 역할을 합니다.
UPR 센서 IRE1, PERK 및 ATF6을 포함한 분자 신호 메커니즘과 신호 캐스케이드 및 XBP1 및 CHOP와 같은 주요 매개체가 설명되었습니다.
수십 년에 걸쳐 비만, 자가면역 질환, 암, 간 질환, 신경퇴행성 질환 등을 포함한 질병에서 ER 스트레스의 역할이 널리 밝혀졌습니다.
ER 스트레스는 염증 촉진 및 항염증 경로를 모두 조절하여 유기체의 항상성을 유지합니다( 그림 3 ).
면역 세포와 ER 스트레스 사이의 상호 관계를 설명하는 데 관심이 높습니다( 표 1 ).
IRE1 경로는 지금까지 가장 많은 관심을 받았고 더 잘 연구되었습니다.
그러나 다중 면역 세포 집단을 조절하는 데 있어 PERK 및 ATF6 경로의 역할은 상대적으로 더 모호합니다.
TME는 스트레스와 적응을 연구하는 유익한 모델입니다.
특히 고형 종양 환경에서 TME는 영양 결핍, 저산소증, 산성 세포외 pH 및 ROS 축적을 특징으로 할 수 있는 종양 세포와 숙주 면역 세포 모두에 적대적인 조건을 만듭니다.
반면에 암세포는 이러한 가혹한 조건에 잘 적응하지만 TME가 면역 회피를 위해 숙주 면역 체계를 다시 배선하기 위해 면역 억제 스트레스 신호를 제공한다는 증거가 증가하고 있습니다
중요하게도, TME의 이러한 모든 대사 조건은 ER 스트레스의 활성제입니다
한편, 유방, 폐, 뇌, 결장, 교모세포종 및 췌장암 세포를 포함하여 빠르게 성장하는 종양 세포 는 표적 치료를 위한 잠재적인 전략을 생성하는 UPR을 활성화하는 것으로 알려져 있습니다.
이러한 노력의 예는 무수히 많습니다. 예를 들어, 우성 음성 IRE1α의 강제 발현 또는 RNA 간섭에 의한 XBP1 억제는 인간 종양 이종이식 모델에서 혈관 형성을 감소시켰습니다
PERK 경로의 억제는 산화적 DNA 손상을 촉진하고 저산소 상태에서 종양 생존을 손상시킵니다
한편, T 세포 항종양 면역과 같은 숙주 면역 반응을 부정적으로 조절하는 ER 스트레스의 새로운 역할이 보고되었습니다.
따라서 ER 스트레스를 표적으로 하는 것은 잠재적으로 암-내인성 및 외인성 경로를 통해 암에 대한 치료가 될 수 있습니다.
표 2에 요약된 바와 같이, 전임상 연구는 면역 세포를 조절하는 UPR 표적화의 가능성을 보여주었습니다.
그러나 이들 제제 중 어느 것도 인간 질병에 대한 임상 승인으로 진행되지 않았습니다.
ER 스트레스를 체계적으로 표적화하면 주로 정상적인 기관 기능에 대한 품질 관리의 손실로 인해 원치 않는 부작용이 크게 발생할 수 있다는 우려가 있습니다.
더욱이, UPR 억제의 유익한 효과와 유해한 효과 사이의 균형을 달성하는 방법과 ER 스트레스의 세포 유형별 표적화가 달성될 수 있는지에 대한 중요한 과제가 남아 있습니다.
ER 스트레스는 종양의 염색질 변화 와 전사체의 조절을 조절하는 중요한 요소입니다 .
따라서 ER 스트레스는 TOX 매개 CD8 + T 세포 고갈 을 조절하는 데 이전에 탐색되지 않은 역할을 할 수 있습니다 .
***************************************************************
2020
UPR(Unfolded protein response) 통합 신호 네트워크는 저산소 상태에서 세포 운명을 결정한다.
https://cmbl.biomedcentral.com/articles/10.1186/s11658-020-00212-1
저산소 스트레스는 세포의 대사 및 혈관신생 경로를 변경하고 산소 항상성을 회복하여 세포가 적응하고 생존하도록 돕기 위해 전반적인 유전자 발현 변화를 유도합니다.
이러한 복구 및 적응 메커니즘이 실패하면 세포는 유전자 발현 프로필을 수정하여 프로그램된 세포 사멸을 유도합니다.
낮은 산소 수준에 대한 대사 적응 및 관련 조직 재혈관화는 대부분의 인간 종양의 생존 및 진행을 허용하고., 황반 변성., 녹내장 진행에 기여합니다..
저산소증에 대한 세포 반응의 주요 목표는 세포 생존을 촉진하고 산소 항상성을 회복하는 것입니다.
불규칙한 단백질 접힘은 또 다른 특정 스트레스 반응 경로인 펼쳐진 단백질 반응(UPR)을 활성화합니다.
UPR은 고유한 신호 전달 네트워크를 통해 소포체 및 미토콘드리아 항상성을 회복하여 세포 생존을 촉진합니다.
그러나 실패하면 UPR은 세포 사멸을 유도합니다.
UPR의 활성화는 생존하는 저산소증을 지원하지만 세포 생존을 손상시킬 수도 있습니다.
예를 들어, ER은 혈관 내피 성장 인자(VEGF)와 같은 혈관신생 수용체 및 리간드를 포함 하는 막횡단 및 분비 단백질의 접힘 및 성숙을 담당하고, erythropoietin(EPO)은 각각 저산소증으로 인한 혈관 신생 및 적혈구 생성에 중요한 역할을 합니다.
저산소증에 대한 저산소증 유발 인자 반응
충족되지 않은 세포 산소 요구량은 저산소증 유발 인자(HIF)라고 하는 특정 전사 인자의 기능적 이종이량체 α/β-소단위 복합체의 축적에 의해 반영됩니다.
HIF는 저산소증 반응 요소(HRE)라고 하는 특정 표적 서열을 포함하는 유전자의 전사 조절을 통해 저산소증에 대한 적응성 및 세포자멸사 반응을 매개합니다.
세포에 산소가 충분히 공급되면 활성 HIF 복합체의 형성은 알파(α) 소단위의 제한된 가용성으로 인해 억제됩니다.
대조적으로, HIF-β 소단위의 세포 수준은 산소와 무관합니다.
저산소 상태에서 HIF-α 소단위의 번역 후 변형이 억제되어 알파 소단위와 전사 활성 HIF-αβ 이종 복합체가 축적됩니다.
HIF-1α가 고등 후생동물에서 HIF 신호전달의 주요 매개체로 간주된다는 사실에도 불구하고, α-소단위체의 다른 조직 특이적 동형인 HIF-2α 및 HIF-3α도 저산소증에 대한 세포 반응에 참여하는 것으로 알려져 있습니다.
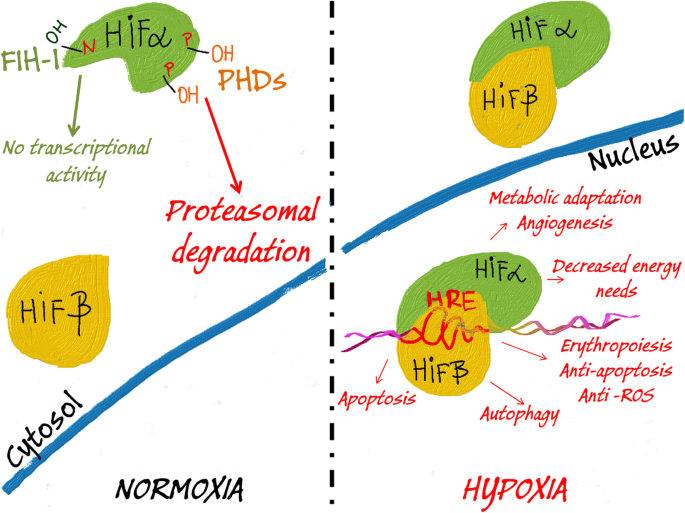
산소 가용성은 HIF 신호를 조절합니다.
정상산소 상태에서 HIFα 서브유닛의 프롤린(P) 잔기는 프로테아좀 분해를 표시하는 PHD에 의해 하이드록실화됩니다.
또한 FIH-1은 HIFα의 아스파라긴 잔기(N)의 하이드록실화를 매개하여 HIF 전사 활성을 방지합니다.
저산소증은 HIFα 소단위를 수산화하는 PHD 및 FIH-1의 능력을 손상시키고, 따라서 이 소단위의 축적 및 안정한 HIFβ 소단위와의 이종이량체화를 초래합니다.
핵에서 HIFα/β 복합체는 HIF 표적 유전자의 HRE 요소에 결합하고 세포를 저산소 조건에 적응시키기 위해 발현을 통제합니다.
생존을 위한 길
저산소 상태에서 HIF는 생존율이 낮은 전사체 전략을 실행하여 세포가 덜 효율적인 비산화 에너지 생산을 통해 에너지 수준을 유지할 수 있도록 합니다.
에너지 수준을 유지하기 위해 HIF는 포도당 수송체 유전자와 해당 효소를 상향 조절하고 산화적 인산화를 억제합니다
(1) 피루브산이 아세틸-Co-A로 전환되는 것을 방지하고
(2) 포도당 산화를 감소
(3) 지방산의 β-산화를 억제
중요하게는, 이 대체 대사 경로의 저산소증 관련 이용은 혐기성 해당 경로의 보다 효율적인 이용을 허용하고 세포에 대한 부정적인 영향을 최소화하는 메커니즘의 HIF 매개 활성화를 동반합니다.
목표는 전자 전달 효율을 높이고 활성 산소 종(ROS) 생성을 줄이는 것입니다.
HIF-1은 또한 (COX) 소단위 구성을 조절하여 저산소 상태에서 호흡 효율을 최적화하고 ROS 소거 경로를 촉진하여 ROS를 감소시킵니다
또한, 혐기성 해당작용은 양성자 방출을 증가시키기 때문에 HIF-1은 탄산탈수효소 9( CA - IX) 의 발현을 유도하고, ( MCT4 )는 세포내 pH를 조절하여 산증을 중화합니다
ATP의 세포 수준의 비산화적 에너지 생산은 산화적 인산화보다 덜 효율적이기 때문에 HIF는 세포의 에너지 요구량을 줄이기 위해 경로를 활성화합니다. 이를 달성하기 위해 HIF는 선택적으로 번역을 억제하여 총 단백질 생산을 감소시키고, 자가포식과 미토 파지를 유도 합니다 .
특히, mTOR 경로는 또한 단백질 합성 및 세포 성장을 감소시키고 HIF-독립적 메커니즘을 통해 자가포식을 유도합니다.
산소 항상성을 회복하고 내피의 웰빙을 유지하기 위해 HIF는vascular endothelial growth factor (VEGF) , heme oxygenase-1 (HMOX1), matrix metalloproteinases (MMP) 2 and 13 , the stem cell factor OCT-3/4, angiopoietin 2 (ANGPT2), stromal derived factor 1 (SDF1) , platelet-derived growth factor B (PDGFB) , placental growth factor (PGF), and stem cell factor (SCF ) and endothelial nitric oxide synthase (NOS3) 을 포함하는 다수의 혈관신생 유전자의 발현을 자극합니다
HIF 유도 혈관신생은 저산소 조직으로의 혈류 증가를 보장하는 반면, 혈액의 산소 관리 능력은 에리트로포이에틴의 HIF 의존적 상향조절을 통해 향상됩니다.
중요한 것은 효율적인 적혈구 생성에 필요한 적절한 세포 철 수준을 확보하기 위해 HIF가 철 항상성을 매개하는 다른 유전자뿐만 아니라 트랜스페린의 발현을 조정한다는 것입니다.
또한 EPO는 항-세포자멸사 단백질을 지원하고 카스파제 활성을 억제합니다.
저산소증에 대한 UPR 경로 반응
저산소증에 대한 세포 반응의 기본 기능은 불안정한 조건에서 생존하고 산소 항상성을 회복하는 것입니다.
따라서, 혐기성 해당과정의 부정적인 영향과 감소된 에너지 가용성을 줄이기 위한 HIF 관련 메커니즘에도 불구하고 이 대사 스위치는 결국 세포 항상성을 방해합니다.
이러한 에너지 결핍은 이온 항상성 유지 및 관련 산화환원 전위와 같은 ATP 의존적 과정의 활성을 제한하고, 손상된 이황화 결합 형성 및 ROS 활성으로 인한 단백질 및 지질 합성, 번역 후 단백질 접힘 능력을 제한합니다.
이러한 모든 요인은 소포체 항상성을 교란할 수 있으며(ER 스트레스라고 함), ER에서 접히지 않거나 잘못 접힌 단백질의 축적으로 이어질 수 있습니다.
잘못 접힌 단백질의 축적은 접힌 단백질 반응(UPR)이라고 하는 또 다른 특수화된 스트레스 반응 신호 전달 경로를 활성화합니다.
저산소 상태에서는 미토콘드리아 기능에 중요한 변화가 있어 ROS 수준이 높아집니다.
또한, 미토콘드리아로 암호화된 단백질의 적절한 접힘과 미토콘드리아 핵으로 암호화된 단백질의 수입 및 상응하는 재접힘은 미토콘드리아 기능에 중요합니다.
따라서 장기간의 저산소증은 결국 미토콘드리아 단백질 접힘의 섭동과 미토콘드리아 전개 단백질 반응(UPRmt)이라고 하는 관련 특정 스트레스 반응 메커니즘의 활성화를 초래할 것입니다.

UPR 및 UPRmt 신호. ER에서 잘못 접힌/펼쳐진 단백질이 축적되면 BIP가 ER 막에서 방출되어 PERK 이합체화 및 후속적인 자가인산화를 유도합니다.
활성화된 PERK는 eIF2α를 인산화하여 전체 번역 감쇠를 유발합니다.
그러나 ATF4를 포함한 일부 전사체는 번역된 상태로 유지되어 있습니다.
ATF4는 ER 항상성을 회복하기 위한 전사 신호를 제공하지만, 이는 또한 proapoptotic CHOP를 유도할 수 있습니다.
유사하게, 미토콘드리아에서 펼쳐진 단백질의 축적은 PERK 활성화 및 ATF4 신호전달(UPRmt)의 유도로 이어집니다.
BIP에서 해리되면 IRE1α는 올리고머화 및 자가인산화를 거쳐 엔도리보뉴클레아제 활성을 얻습니다.
ER 부하를 줄이기 위해 활성화된 IRE1α는 mRNA와 miRNA(RIDD)를 분해합니다.
IRE1α는 또한 XBP1 mRNA의 스플라이싱을 수행하여 전사적으로 활성인 XBP1을 방출합니다.
XBP1s는 ER 항상성을 회복하기 위해 전사 프로그램을 활성화합니다.
대안적으로, IRE1α는 proapoptotic kinase JNK1을 활성화할 수 있습니다.
마지막으로, BIP 해리는 ATF6이 골지로 이동하는 것을 허용하며, 여기서 이 단백질의 절단은 전사적으로 활성인 ATF6f의 방출을 초래합니다.
ATF6f는 전사 프로그램을 활성화하여 ER 항상성을 회복하고 ERAD를 지원합니다
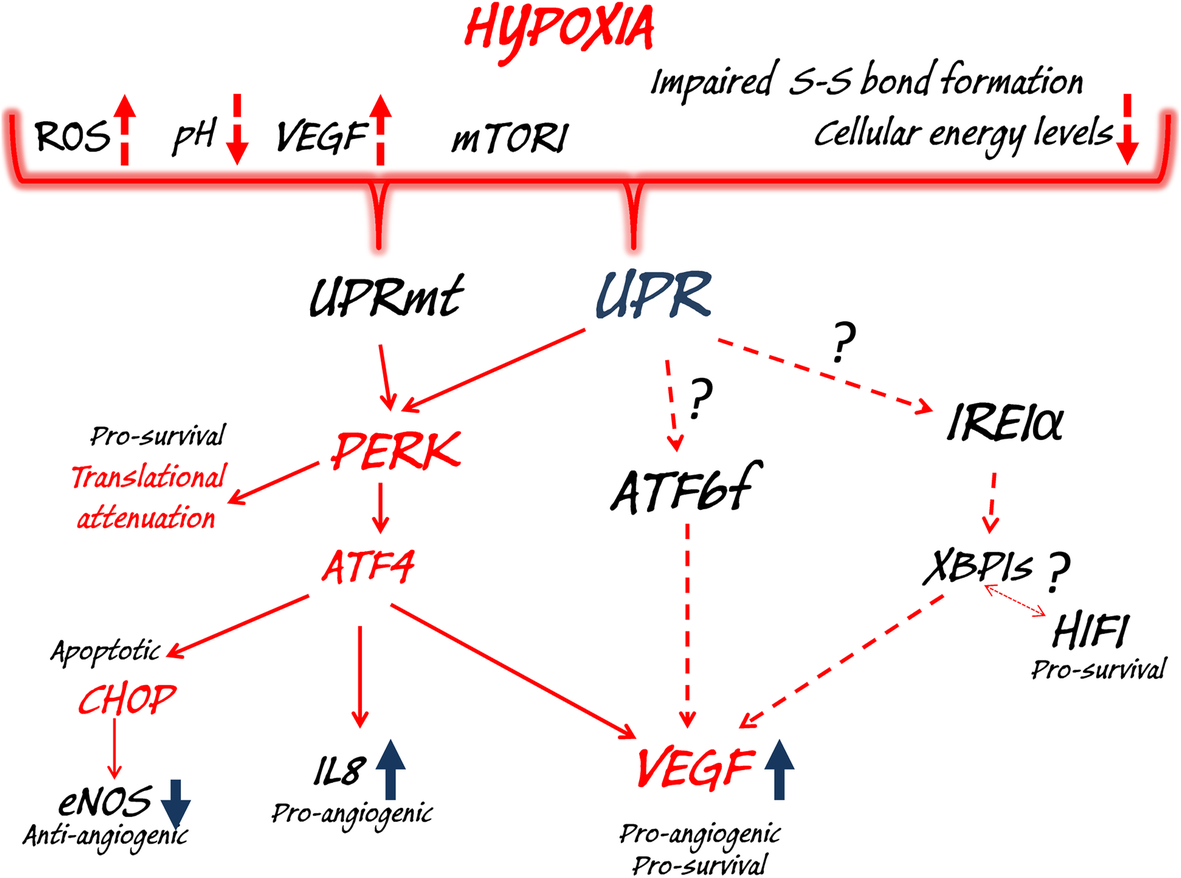
저산소증 신호 및 세포 기능의 관련 변화는 UPR 및 UPRmt를 활성화합니다.
저산소 상태에서 ER과 미토콘드리아에 잘못 접힌/펼쳐진 단백질이 축적되면 PERK 신호전달이 활성화되고, 이는 생존(전역적 번역 정지-global translational arrest 및 혈관신생 촉진 유전자 IL8 및 VEGF의 유도)과 세포자멸사 반응(CHOP의 유도 및 친혈관형성 eNOS 발현), 또한, 일부 모델에서 ATF6 및 IRE1α의 저산소증 관련 활성화는 생존 및 혈관 신생 신호 전달에 기여합니다.
또한 생존 신호에서 XBP1과 HIF1 사이에 협력이 있는 것으로 보입니다.
종합적으로, 암세포에서 보고된 다양한 ER 스트레스 관련 메커니즘에도 불구하고 저산소증에 노출되면 주로 UPRmt에 의해 공동 조절될 수 있는 PERK 축의 활성화가 발생합니다
이에 비해 중등도 및 장기간의 저산소 상태 동안 UPR 및 UPRmt의 제한된 활성화는 HIF가 광범위한 ROS 형성을 성공적으로 방지하고 스트레스 상태를 완화함을 시사합니다.
이 진술은 저산소증에 노출된 HIF-1α 녹아웃 세포가 치명적인 수준의 ROS를 생성한다는 연구 결과와 세포 ROS 수준과 HIF-1 안정화 사이의 음의 상관 관계를 보여주는 수많은 보고서에 의해 뒷받침됩니다.
그러나 놀랍게도 누적된 증거에 따르면 저산소 세포에서 정상적인 산소 수준이 빠르게 재확립되면 종종 광범위한 ROS 생성이 발생하고 저산소증-재산소화 손상 및 허혈-재관류 손상이라고 하는 세포 손상이 발생합니다. ROS는 저산소 세포에 축적되지만 산소의 빠른 재도입으로 인해 그 수준이 극적으로 증가합니다
광범위한 UPR 및 UPRmt 활성화는 인간 내피 세포, 심근세포 및 뉴런에서 산소 수준을 빠르게 회복할 때 발생했습니다.
중요하게도, 간헐적(주기적 저산소증)은 저산소증/재산소화 주기에 대한 세포의 만성 노출로 인해 발생하며 수면 무호흡증의 기본 특징입니다.
주기적인 저산소증은 또한 만성 ER 스트레스를 동반하는 것으로 밝혀진 대부분의 고형 종양의 발달을 명확하게 규정합니다.
고형 종양은 만성 저산소증보다는 변동하는 산소 수준(주기적 저산소증)에 지속적으로 노출됩니다
그럼에도 불구하고, 저산소증으로 유발된 UPR 암 연구의 대부분은 감소된 UPR 활성화를 나타내는 연속 저산소증 모델에서 수행되었습니다.
********************************************************
2018
소포체 스트레스 신호 - 기본 메커니즘에서 임상 적용에 이르기까지
--가장 자세한 설명--읽어보세요
FEBS Press
The current article reviews the most up-to-date literature on Endoplasmic Reticulum (ER) biology and articulates this information from a signalling perspective. Not only do we cover the basic cell bi...
febs.onlinelibrary.wiley.com
Table 1. UPR-변환기 단백질 경로를 표적으로 하는 다양한 조절제
| PERK | GSK2656157 | PERK Kinase | In preclinical stage for multiple myeloma and pancreatic cancer | |
| Salubrinal | GADD34/PP1c | Inhibition of eIF2α dephosphorylation | ||
| In ALS, it increases lifespan of mutant superoxide dismutase 1 transgenic mice | ||||
| In Parkinson's disease, it increases neuronal survival of α-synuclein transgenic mice | ||||
| ISRIB | eIF2β | Decreased ATF4 expression | ||
| Guanabenz | GADD34/PP1c | Inhibitor of eIF2α phosphatase, | ||
| Sephin1 | GADD34 (PP1c) | Inhibitor of eIF2α phosphatase | ||
| IRE1 | Salicylaldimines | IRE1 RNase | IRE1αRNase active-site inhibitor | |
| STF-083010 | IRE1 RNase | IRE1α RNase active-site inhibitor | ||
| In preclinical stage for multiple myeloma treatment | ||||
| MKC-3946 | IRE1 RNase | IRE1α RNase active-site inhibitor | ||
| In preclinical stage for multiple myeloma treatment | ||||
| 4μ8c | IRE1 RNase | IRE1α RNase active-site inhibitor | ||
| In preclinical stage for multiple myeloma treatment | ||||
| APY29 | IRE1 Kinase | IRE1α kinase active-site inhibitor | ||
| Sunitinib | IRE1 Kinase | IRE1α kinase active-site inhibitor | ||
| FDA approved for renal cell carcinoma | ||||
| It acts on multiple kinases | ||||
| KIRA | IRE1 Kinase | IRE1α kinase active-site inhibitor | ||
| Toyocamycin | IRE1 RNase | IRE1α RNase active-site inhibitor | ||
| In preclinical stage for various cancers treatment | ||||
| 3-ethoxy-5,6-dibromosalicylal- dehyde | IRE1 RNase | IRE1α RNase active site inhibitor | ||
| Apigenin | Proteasome | Increase of IRE1a nuclease activity in model | ||
| FIRE peptide | IRE1 Kinase | Modulation IRE1 oligomerization in vitro, | ||
| Xbp1 mRNA cleavage in vitro, in cell culture and in vivo (Caenorhabditis elegans) | ||||
| ATF6 | Apigenin | ATF6 | Upregulation of ATF6 expression | |
| Baicalein | ATF6 | Upregulation of ATF6 expression | ||
| Ceapin | ND | Inhibitor of ATF6 | ||
| Kaempferol | ATF6 | Downregulation of ATF6 expression | ||
| Melatonin | ATF6 | Inhibitor of ATF6 | ||
| Compound 147 | ATF6 | Activator of ATF6 | ||
| Compound 263 | ATF6 | Activator of ATF6 | ||
| 16F16 | PDI | Inhibitor of PDI |
***************************************************************
2021
소포체 스트레스와 유기체
https://j-organoid.org/journal/view.php?doi=10.51335/organoid.2021.1.e3
Endoplasmic reticulum stress and organoids
Introduction Organoids are characterized by three-dimensional (3D) epithelial primary cell cultures that develop and self-organize from stem cells via cell sorting and spatially restricted descendant commitment, similar to the in vivo process [1]. Embryoni
j-organoid.org
ER 스트레스를 표적으로 하는 암 치료제 개발에 오르가노이드 적용
Compound name ER Organoid applied Cancer Reference
| 1 | Curcumin | ↑ IRE1α, ATF6, caspase-12 | No | Prostate, myeloma | Huang et al. [44], Rivera et al. [45] |
| 2 | Curcumin + oligomeric proanthocyanidins | ↓ GRP78, HSPA5 | Yes | Colorectal | Ravindranathan et al. [46], Toden et al. [47] |
| 3 | Curcumin + sildenafil | ↑ p-eIF2α, CHOP | No | Gastric, colon, liver | Roberts et al. [48] |
| 4 | Curcumin + irinotecan | ↑ CHOP, PDI, BiP | No | Colorectal | Huang et al. [49] |
| 5 | Bisdemethoxycurcumin, demethoxycurcumin | ↑ BiP, IRE1α, CHOP, ATF6, caspase-4 | Yes | Lung, colorectal | Ko et al. [50], Wang et al. [51], Yang et al. [5 |
| 6 | B63 (curcumin analogue) | ↑ ER stress markers | No | Colon | Zheng et al. [53] |
| 7 | B19 (curcumin analogue) | ↑ ROS, p-PERK, p-eIF2α, CHOP | No | Ovarian | Qu et al.[54] |
| 8 | WZ35 (curcumin analogue) | ↑ p-eIF2α, CHOP, ATF4 | No | Breast, liver, prostate | Wang et al. [55], Wang et al. [56], Zhang et |
| 9 | MTH-3 (curcumin analogue) | ↑ CHOP, ERO1, PDI, PERK | No | Breast | Chang et al. [58] |
| 10 | EGCG | ↑ Binding to BiP | No | Bladder | Luo et al. [59] |
| 11 | JP8 (EGCG analogue) | ↑ ATF4, CHOP | No | Melanoma | Xie et al. [60] |
| 12 | Polyphenon E® | ↑ CHOP | No | Prostate | Rizzi et al. [61] |
| 13 | γ- Tocotrienol | ↑ BiP, p-PERK, p-eIF2α, ATF4, CHOP, p-IRE1α, caspase-12 | No | Breast, cervical, melanoma | Montagnani Marelli et al. [62]; Park et al. [6 |
| 14 | δ- Tocotrienol | ↑ BiP, CHOP, PERK, IRE1α, p-eIF2α, ATF4 | No | Melanoma | Montagnani Marelli et al. [62] |
| 15 | Pimpinelol | ↑ ATF4, CHOP, GADD34 | No | Breast | Aghaei et al. [65] |
| 16 | Pristimerin | ↑ ATF4, CHOP, IRE1α, p-eIF2α | Yes | Breast, lung | Tang et al. [35], Cevatemre et al. [66] |
| 17 | Cnidium officinale | ↑ ATF4, CHOP | No | Lymphoma | Cha et al. [67] |
| p-eIF2α, p-PERK | |||||
| 18 | Salvia miltirrhiza | ↑ ATF4, CHOP | No | Lymphoma | Kim et al. [68] |
| p-eIF2α, p-PERK | |||||
| 19 | Protodioscin | ↑ ATF4, CHOP | No | Cervical | Lin et al. [69] |
| p-eIF2α, p-PERK, JNK, BiP | |||||
| 20 | Paenia suffruticosa | ↑ DAPK3 | No | Pancreas, liver | Liu et al. [70] |
| 21 | Clinacanthus nutans | ↑ IRE1α, CHOP | No | Lymphoma and leukemia | Lu et al. [71] |
| 22 | Chrysophanol | ↑ ROS, p-PERK, p-eIF2α, CHOP | No | Breast | Park et al. [72] |
| 23 | Garlic extract | ↑ BiP, MAPK kinase, SKP1 | No | Myeloma, prostate | Park et al. [72] |
| 24 | Ajoene | ↑BiP, CHOP | No | Breast | Siyo et al. [73] |
| 25 | 7-Acetylsinumaximol B | ↑ p-PERK, p-eIF2α, ATF4, CHOP, ATF6 | No | Gastric | Tsai et al. [74] |
| 26 | 4-Nerolidylcatechol | ↑ p-PERK, IRE1α, BiP, CHOP | No | Melanoma | Alves-Fernandes et al. [75] |
| 27 | PP-22 | ↑ PERK, CHOP, BiP, PDI | No | Nasopharyngeal | Tan et al. [76] |
| 28 | Mangosteen fruit extract | ↑ BiP, PERK, IRE1α calnexin, CHOP, caspase-4 | No | Prostate | Li et al. [77] |
| 29 | Garcinone-E | ↑ BiP, IRE1α, XBP1, CHOP, caspase-12 | No | Ovarian | Xu et al. [78] |
| 30 | Gartanin | ↑ CHOP | No | Prostate | Li et al. [79] |
| 31 | Garcinol | ↑ CHOP | No | Liver | Cheng et al. [80] |
| 32 | Gambogic acid | ↑ BiP, XBP1s, CHOP, GADD34, JNK | Yes | Cervical, prostate | Cheng et al. [80], Pan et al. [81] |
| 33 | Resveratrol | ↑ IRE1α, CHOP, JNK activation, ATF6α, PERK, p-eIF2α, BiP | No | Myeloma, lung, nasopharyngeal, ovarian | Bai et al. [82], Chow et al. [83] Gwak et al. |
| 34 | GSK2656157 | ↓ PERK kinase | No | Colorectal | Zheng et al. [53], Axten et al. [86] |
| 35 | GSK2606414 | ↓ PERK kinase | Yes | Head and neck | McLaughlin et al. [87] |
| 36 | Salubrinal | ↑ eIF2α | Yes | Breast, ovary, prostate | Bastola et al. [88], Jeon et al. [89], Zadra et |
| 37 | ISRIB | Inhibition of p-eIF2α | Yes | Prostate | Nguyen et al. [91] |
| 38 | Sephin1 | Inhibition of p-eIF2α | Yes | Brain | Pellegrini et al. [92] |
| 39 | Salicylaldimines | Inhibition of IRE1α RNase | No | Liver | Jakku et al. [93] |
| 40 | STF-083010 | Inhibition of IRE1α RNase and XBP1 | No | Breast, pancreatic, ovarian | Barez et al. [94], Ming et al. [95], Thakur et a |
| 41 | MKC-3946 | Inhibition of IRE1α RNase | No | Myeloma, pancreatic, leukemia | Lugea et al. [97], Mimura et al. [98], Sun et |
| 42 | 4µ8C | Inhibition of IRE1α RNase | Yes | Colon | Li et al. [23] |
| 43 | Sunitinib | Inhibition of IRE1α kinase | No | Pancreatic | Thakur et al. [96] |
| 44 | Toyocanamycin | Inhibition of XBP1 mRNA Splicing | Yes | Myeloma | Joly et al. [100] Ri et al. [101] |
| 45 | 3-Ethoxy-5,6-dibromosalicylaldehyde | Inhibition of XBP1 splicing | No | Pancreatic | Chien et al. [102], Volkmann et al. [103] |
| 46 | Apigenin | ↑p-IRE1α, p-eIF2α, ROS | Yes | Colorectal, breast | Park et al. [104], Xu et al. [105] |
| 47 | Baicalein | ↑ ATF6 | No | Liver, breast | Wang et al. [106], Yan et al. [107] |
| 48 | Ceapin | Inhibitor of ATF6 | No | Liver | Torres et al. [109] |
| 49 | Kaempferol | ↓ ATF6 | No | Gastric | Kim et al. [109] |
| 50 | Melatonin | Inhibition of ATF6 | Yes | Colorectal | Sakatani et al. [43], Lee et al. [110] |
| 51 | 16F16 | PDIA3 inhibitor | No | Breast | Young et al. [111] |
| 52 | ONC201 | Inhibit eIF2α | No | Breast | Yuan et al. [112] |
| 53 | Eeyarestatin I (EerI) | ERAD inhibitor | No | Blood | Wang et al. [113] |
↑: upregulation; ↓: downregulation.
ATF, activating transcription factor; BiP, CHOP, DAPK3, eIF2α, ER, ERAD, ERO1, GADD34, GRP78, HSPA5, IRE1α, XBP1, JNK, MAPK, PDI, PDIA3, PERK, ROS, SKP1, SN, XBP1.
**********************************************************
https://www.frontiersin.org/articles/10.3389/fcell.2021.683940/full
Endoplasmic Reticulum Stress and Tumor Microenvironment in Bladder Cancer: The Missing Link
Bladder cancer is a common malignant tumor of the urinary system. Despite recent advances in treatments such as local or systemic immunotherapy, chemotherapy, and radiotherapy, the high metastasis and recurrence rates, especially in muscle-invasive bladder
www.frontiersin.org
******************************************************************
'암치료' 카테고리의 다른 글
| 암 치료의 식물: 전임상에서 임상 까지 (0) | 2022.01.10 |
|---|---|
| 자연치유 암-항종양 효능이 있는 천연 활성 화합물 (0) | 2022.01.10 |
| 암 환자에서 보완 대체 의약품의 잠재적인 위험 (0) | 2022.01.07 |
| 4/4) 암과 해를 끼칠 수 있는 천연 제품 (0) | 2022.01.07 |
| 3/4) 암 성장을 방지하는 천연 제품 (0) | 2022.01.07 |