https://cafe.naver.com/starvecancer/1598
암에서 AMPK 신호전달 경로의 역할
대한민국 모임의 시작, 네이버 카페
cafe.naver.com
AMPK(5′-AMP-activated protein kinase)는 1987년 지방산 및 콜레스테롤 합성을 조절하는 인산화 효소로 발견 된 이후 , 현재 다양한 대사 스트레스 상황에서 활성화 되어 대사과정 을 조절함으로써 에너지 및 레독스(redox) 항상성을 유지하는데 있어 매우 중요한 역할을 수행한다.
대사 스트레스 상 황에서 활성화 된 AMPK는 단백질 및 지방산 합성과 같이 ATP 및 NADPH를 소모하는 과정을 차단하고, 지방산 분 해와 같이 이들을 생산하는 과정을 활성화시킴으로써 에너 지 및 레독스 항상성을 유지하게 되고 궁극적으로 세포의 생 존 및 사멸을 조절한다.
이처럼 AMPK는 대사 조절 과정에 서 중요한 역할을 담당하고 있어 주요 대사 질환인 암 및 당 뇨의 발병 기전을 이해하고 치료전략을 수립하는데 있어 중 요하게 여겨지고 있다.
흥미롭게도, 특히 암에서 지금까지 진행되어 온 AMPK의 역할에 관한 연구 결과를 보면 단순히 암 유발 또는 억제 인자로서 단정 지을 수 있기보다는 양날의 칼처럼 두 가지 측면을 모두 가지고 있음을 알 수 있다.
암에서 AMPK의 활성화 조절
AMPK는 에너지 고갈, 세포외 기질 이탈, 활성 산소 증가 및 저산소 상황과 같은 다양한 대사 스트레스에 의해 활 성화되어 대사 적응(adaptation)을 유도하는데 중요한 역 할을 수행한다.
흥미로운 점은, 바로 이러한 스트레스 상황 이 모두 종양 미세환경에서 공통적으로 발생하는 현상이라 는 점이고 최근에 뇌종양 조직에서 실제로 AMPK가 활성 화 되어 있음이 보고된 바 있다.
에너지 고갈로 인한 AMPK 활성화 기전은 비교적 잘 알려져 있으며 이는 LKB1(Liver Kinase B1)에 의한 AMPK의 인산화로 활성화 되는 것 등 으로 제시되고 있다.
세포외 기질 이탈에 의한 AMPK 활 성화는 최근에 보고가 되었으며 이는 LKB1 뿐만 아니 라 CaMKK2(Calcium/calmodulin-dependent Protein Kinase Kinase 2) 에 의한 인산화로 활성화 되는 것으로 제 시되고 있다.
활성 산소 증가 및 저산소에 의한 AMPK 활성 화는 알려진지 오래 되었음에도 불구하고 그 기전에 대해서 는 아직 논란의 여지가 있다.
이러한 종양 미세환경의 요소 들뿐만 아니라 최근에는 암에서 발견되는 종양유발 유전자의 활성화 및 억제 유전자의 비활성화에 의해서도 AMPK 가 활성화 된다고 밝혀지고 있지만 이 역시 자세한 기전은 분명치 않다. 그렇다면 여기서 중요한 질문은 암세포에서 활성화된 AMPK의 역할이 과연 종양 억제인지 아니면 촉 진인지에 관한것으로 이와 관련된 최근 연구 결과들에 대 해 각각 논의해 보고자 한다.
종양 유발 억제 인자로서의 기능
초기에 제시된 암에서의 AMPK의 역할은 종양 유발 억제 기능에 초점을 두고 있다.
이는 2003~2004년 종양 유발 억제 인자로 잘 알려진 LKB1이 AMPK의 주요 인산화 효소 로 밝혀짐과 동시에 AMPK가 TSC2(Tuberous Sclerosis Complex 2)를 통해 mTORC1(mechanistic Target of Rapamycin Complex 1)을 억제한다는 것이 밝혀지면서 부 터라고 할 수 있다.
즉, AMPK가 LKB1에 의한 mTORC1의 억제를 매개한다는 사실이 밝혀지면서 자연스럽게 AMPK 가 종양 유발 억제 인자로서 역할을 하게 될 것이라는 가설 을 세우게 되었고, 그 후 AMPK의 활성화를 유발하는 것으 로 알려진 당뇨병 치료제인 metformin 복용 환자들을 대상 으로 한 후향적 역학조사를 통해 metformin 복용과 암 발 생 사이에는 실제로 역상관 관계가 있음이 제시되었다.
또 한 in vitro 및 in vivo 실험을 통해 metformin 뿐만 아니 라 phenformin, AICAR 등 AMPK 활성화를 유도하는 여 러 가지 약물을 처리한 경우 암세포의 성장 및 생존이 억제 됨을 실험적으로 증명하게 되었다.
이처럼 비교적 풍부한 약물학적 증거와는 달리, AMPK가 종양 유발 억제 인자임 을 뒷받침하는 유전학적 증거는 최근에 이르러서야 캐나다 의 Jones 그룹에 의해 제시되었다.
이들은 유전자 조작 마 우스모델을 이용하여 c-myc 발현에 의한 림프종 유발 과정 에서 AMPKα1이 결손 되었을 때 mTORC1의 활성화와 함 께 발암이 촉진됨을 확인함으로써 AMPK가 실제 암의 유 발에 있어서 억제 인자로 작용한다는 증거를 제시하게 되었 다.
하지만 이 결과는 최근에 발표된 c-myc 발현에 의한 간암 유발에서 AMPK가 필수적임을 보여준 다른 그룹의 연 구 결과와 배치되는 것이어서 좀 더 세밀한 분석과 심화 연구가 필요하다고 하겠다.
종양 유발 촉진 인자로서의 기능
그동안 제시되었던 그 약물들에 의한 암세포의 성장 및 생존 억제 효과는 AMPK의 활성화와는 무관하며, 오히려 AMPK 활성화는 이들에 의한 세포독성으로부터 보 호하는 작용을 한다는 것을 의미하는 것이다.
실제로 최근 직접적으로 AMPK를 활성화 시키는 약물로 찾은 A769662 를 세포에 처리하였을 경우 세포독성은 미미하였으며 오히려 에너지 스트레스 상황에서는 세포 성장을 촉진시킨다는 결론을 얻게 되었다.
종양 유발 촉진 인자로서의 작용 기전
AMPK가 종양 유발을 촉진하는 기전으로는 최근까지 AMPK의 잘 알려진 기능, 즉 세포내 에너지(ATP)의 고갈 을 차단함으로써 대사 스트레스 상황에서 암세포의 생존 을 촉진시키는 것과 연관이 있을 것으로 여겨져 왔다.
이러 한 견해를 뒷받침하는 최근 연구 결과들 중에서, myc 또는 AR(androgen receptor) 활성화에 의한 간암, 또는 전립선 암의 발생과정에서 AMPK에 의한 해당과정 또는 미토콘드 리아 대사의 활성화가 필수적임을 제시한 논문이 발표된 바 있다.
또한 종양억제 유전자로 알려진 folliculin의 돌연변 이에 의한 발암과정에서 AMPK가 활성화 되고 이는 PGC1α -HIF1α의 활성화를 통해 해당과정을 증가시키는 것이 관찰 되었다.
이러한 최근 연구에서 제시된 기전은 비록 AMPK 에 의한 대사의 활성화이긴 하지만, 여전히 ‘ATP’의 생성이 중요한가에 대한 의문의 여지는 남아있다.
흥미롭게도, 최근 미국의 Hay 그룹에서는 AMPK가 ATP 보다는 NADPH 와 GSH의 균형을 유지하여 활성 산소의 양 을 줄임으로써 암세포의 생존 및 성장을 촉진시킨다는 새로 운 결과를 제시하였다.
이 논문에서는 종양 미세환경 인자들 중 두 가지(저 포도당 상태와 세포외 기질 이탈 상태)를 이용 하여 대사스트레스를 유발하였고 이 상황에서 AMPK가 적 절히 활성화 되지 않으면 NADPH 고갈로 인한 산화적 스트 레스에 의해 세포 사멸이 증가함을 보여주었다.
이 상황에서 AMPK가 활성화 되면 지방산의 합성을 억제시키는 반면 산화를 촉진시켜, 각각 NADPH의 소모를 줄이고 합성을 증가 시킴으로써 레독스 균형을 유지하여 암세포의 생존 및 성장 을 촉진시킨다는 것을 제시하였다.
이와 비슷하게 최근 다른 그룹에서도 AMPK가 NADPH를 유지함으로써 산화적 스트 레스에 의한 암세포의 사멸을 감소시킨다는 것이 제시되었 으며 또한 folliculin의 결손에 의한 AMPK 활성화가 활성 산소에 대한 저항성을 증가시켜 세포의 생존을 증가한다는 연구 결과도 제시되었다.
임상적 적용 및 향후 전망
10여 년 전 LKB1-AMPK-mTORC1 신호전달 과정이 발견된 이래로 그동안 AMPK 활성화를 통한 항암 전략이 대세를 이루어 왔던 것이 사실이다.
하지만 앞에서 언급하였듯 이 AMPK의 활성화는 암세포의 성장 및 생존을 촉진시킨다 는 사실이 밝혀졌고, 대사 스트레스를 유발하는 biguanides 계열의 약물에 의한 세포 독성 효과는 AMPK 유전자가 결손 된 세포에서 훨씬 강력하게 나타난다는 연구 결과가 나옴으 로 인해 최근에는 오히려 AMPK 활성의 억제를 통한 항암 전략이 설득력을 얻고 있다.
현재 임상에서 항암제로 사용되고 있는 약물 중 sunitinib이 AMPKα에 직접적으로 결합함으로써 compound C 보다 우수한 AMPK 억제 작용을 나타냄을 보 고하였다.
***********************************************************************************************************
https://aacrjournals.org/mcr/article/13/7/1059/89572/Dissecting-the-Dual-Role-of-AMPK-in-Cancer-From
Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies
Abstract. The precise role of 5′AMP-activated kinase (AMPK) in cancer and its potential as a therapeutic target is controversial. Although it is well established that activation of this energy sensor inhibits the main anabolic processes that sustain canc
aacrjournals.org
Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies
암에서 AMPK의 이중 역할 분석: 실험에서 인간 연구까지
AMPK의 활성화가 암세포 증식과 성장을 유지하는 주요 동화 과정을 억제한다는 것은 잘 확립되어 있지만, AMPK 활성화는 일반적으로 관찰되는 저산소증 및 포도당 결핍과 같은 대사 스트레스 하에서 생존할 수 있는 가소성을 암세포에 부여할 수 있습니다.
AMPK는 "조건부" 종양 억제인자 및 "상황적" 종양유전자로 지칭됩니다.
인간 암 조직의 AMPK 활성화와 종양 공격성 및 진행과의 상관 관계는 상황에 따라 다양한 것으로 보입니다.
5′ AMP-activated kinase(AMPK)는 대사 네트워크와 신호 네트워크 사이의 교차점에 서 있는 중앙 대사 센서입니다.
2003년에 AMPK의 주요 업스트림 키나제로서 종양 억제인자 liver kinase B1(LKB1)의 발견은 에너지 조절제와 암 발병 사이의 연관성을 확립했으며, 이는 LKB1의 종양 억제인자 기능이 AMPK에 의해 매개될 수 있음을 시사합니다 .
암에서 AMPK 활성화의 기능적 결과는 처음 생각했던 것보다 훨씬 더 복잡한 것으로 보이며 AMPK는 상황에 따라 암 "친구" 또는 "적"으로 행동할 수 있습니다.
AMPK의 약물 유도 생리학적 활성화는 주요 생합성 경로의 억제를 통해 시험관 내 및 전임상 모델에서 종양 성장을 감소시킵니다(참고문헌 4, 5 참조).
그러나 광범위한 스트레스(예: 저산소증, 포도당 결핍 및 기질 분리)에 대한 AMPK의 생리학적 활성화는 암세포에 대사 스트레스에 적응하고 생존할 수 있는 유연성을 제공합니다(대사 적응, 참조 6 에서 검토 ).
AMPK 활성화 수준이 다른 종양 유형에서 이질적이라는 것을 밝혀냈지만, AMPK 활성화와 종양 예후 사이의 상관 관계에 대해 불일치 데이터가 보고되었습니다.
AMPK: 다면발현성 다운스트림 표적이 있는 고유한 대사 "보호자"
AMPK는 ATP 생산을 억제(예: 저산소증, 포도당 결핍, 비구아나이드 약물 또는 생체이물 치료)하거나 소비를 가속화하여 ATP 수준을 감소시키는 대사 스트레스 조건에서 세포 및 유기체 수준에서 에너지 항상성을 복원하는 에너지 센서의 기능을 하여 (예: 근육 수축), 결과적으로 ADP 및 AMP 수준의 증가시킵니다.
LKB1은 에너지 스트레스 동안 AMPK를 활성화하는 반면 CaMKK2 활성은 세포 의 에너지 상태에 관계없이 증가된 세포 내 Ca2 + 수준에 의해 유도됩니다(참조 13 참조 ).
그러나 CaMKK2는 AMPK 인산화를 매개하는 LKB1의 부재를 보완할 수 있습니다.
AMP, ADP 및 Ca2 + 외에도 최근 연구에서는 활성 산소종(ROS)이 LKB1 독립적인 방식으로 작용하는 AMPK의 추가 상류 활성제로 확인되었습니다( 그림 1 , 참조 16 ).).
활성화되면 AMPK는 ATP와 NADPH를 소비하는 동화 경로를 차단하여 에너지 균형을 유지하는 반면 대사 효소의 직접적인 인산화와 전사 인자 및 보조 활성화제의 인산화에 의해 매개되는 장기적 효과를 통해 ATP를 생성하는 이화 경로를 전환합니다 .
따라서 AMPK는
(i) 라파마이신 복합체 1(mTORC1) 신호 전달 구성원인 결절성 경화증 복합체 2( tuberous sclerosis complex 2=TSC2) 및 랩터의 포유류 표적의 직접적인 인산화를 통해 단백질 합성을 억제하고,
(ii) 증식하는 세포에서 새로운 막 형성에 필요한 지방산(FA) 및 콜레스테롤 생합성 [효소 아세틸-CoA 카르복실라제 1(ACC1) 및 3-하이드록시-3-메틸글루타릴-CoA 환원효소(HMGR)의 직접적인 인산화 및 지방 생성 전사 인자의 억제를 통한 스테롤 조절 요소 결합 단백질(SREBP) 및 탄수화물 -반응성 요소 결합 단백질(ChREBP)]을 차단함으로써 세포 성장을 억제할 수 있습니다.
(iii) 세포 주기 정지 및 세포자멸사 유도 [p53의 안정화, 사이클린 의존성 키나제 억제제 p21 Waf1 의 조절을 포함한 여러 메커니즘 및 그리고 p27Cip1 , 하마 신호전달 구성원 안지오모틴-유사 1(AMOTL1)의 인산화, 예-연관 단백질(YAP)의 업스트림 억제제을 통해 ],
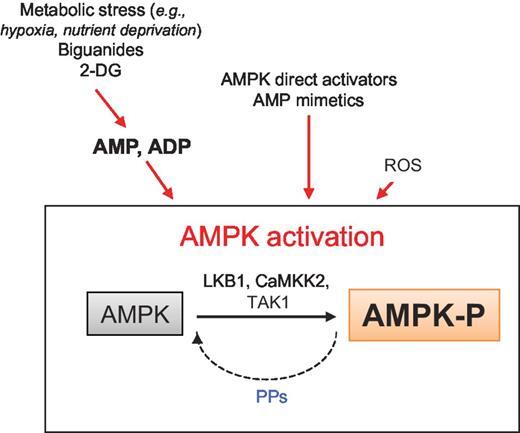
AMPK 활성화 메커니즘.
AMPK는 저산소증, 영양소 결핍 및 약물/화합물[예: 비구아니드, 2-데옥시글루코스(2-DG)], AMP 모방체, 직접 AMPK 활성화제 또는 ROS에 의해 유도된 대사 스트레스에 의해 활성화되는 대사 센서로 기능합니다.
키나제의 완전한 활성을 위해서는 촉매 루프의 residue=잔기 Thr172에서의 인산화가 필요합니다.
주요 업스트림 키나제는 LKB1, CaMKK2 및 TAK1입니다. 특성화되지 않은 protein phosphate(PP)은 이 인산화를 역전시킬 수 있습니다.
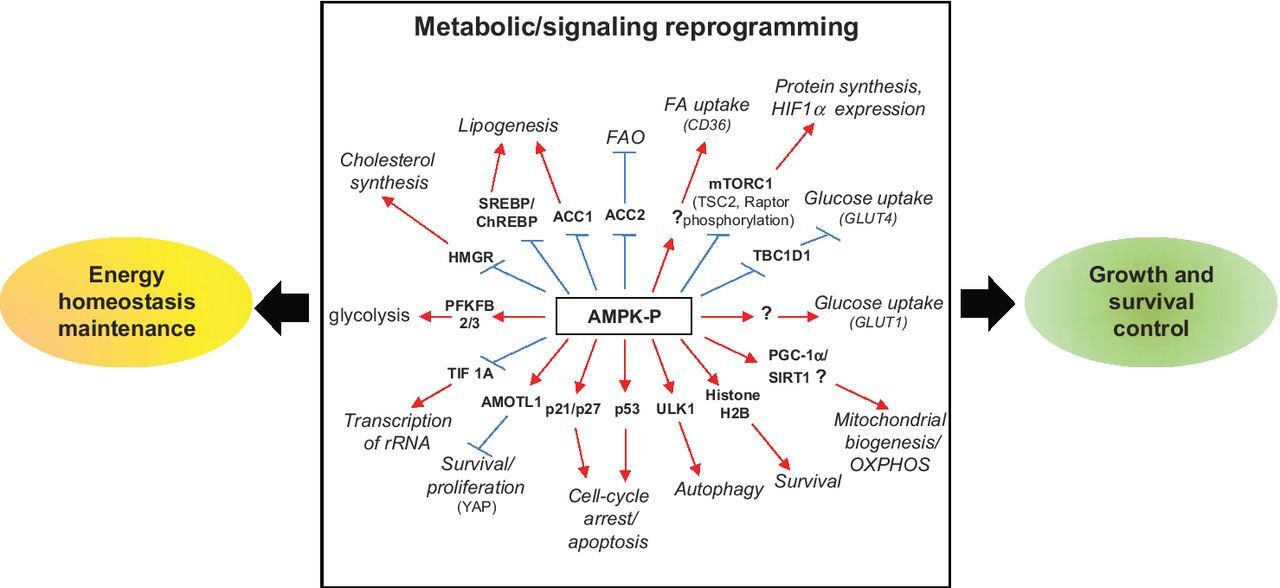
AMPK 매개 대사 및 신호 재프로그래밍.
활성화되면 AMPK는 동화 경로를 끄고 이화 경로를 켜서 에너지 항상성을 복원합니다.
따라서 AMPK는 대사, 세포 성장 및 생존과 관련된 경로를 제어합니다.
빨간색 선은 직접 활성화를 나타내고 억제는 파란색으로 표시됩니다.
물음표는 단백질이 직접 인산화되었는지가 아직 확실하지 않음을 나타냅니다.
GLUT1/4, 포도당 수송체 1, 4; PFKFB2/3,6-포스포프룩토-2-키나제/프룩토스-2,6-비스포스파타제 2 및 3; TBC1D1, TBC1 도메인 단백질-1; SIRT1, 시르투인 1; PGC-1α, PPARγ-공활성화제-1α.
GLUT1/4, glucose transporter 1, 4; PFKFB2/3,6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatases 2 and 3; TBC1D1, TBC1 domain protein-1; SIRT1, sirtuin 1;
암에서 AMPK의 역할: 전임상 연구
종양 억제제로서의 AMPK
종양 억제인자로서의 LKB1의 역할이 잘 확립되어 있기 때문에 AMPK는 주로 LKB1 매개 종양 억제인자 캐스케이드의 구성 요소로 간주되었으며 암에서 자체의 독립적인 역할에 대해서는 훨씬 덜 알려져 있습니다.
이는 대부분의 데이터가 AMPK와 무관한 메커니즘을 나타내는 AMPK 활성화제 AICAR 및 메트포르민을 사용하여 생성되었거나 LKB1 비활성화 모델의 실험적 증거에 의해 생성되었다는 사실 때문이며, 이는 AMPK 외에 추가로 12개의 AMPK 관련 다운스트림 키나제에 영향을 미칩니다.
AMPK 종양 억제 활성의 토대 메커니즘은 반대로 AMPKα1 KO쥐에서 상향 조절되는 저산소증 유발 인자 1-알파(HIF1α)와 그 다운스트림 해당 유전자를 하향 조절함으로써 "항-바르부르크" 효과를 발휘하는 키나제의 능력입니다.
Warburg 효과를 길항하는 것 외에도 AMPK는 G 1 –S 및 G 2 –M 단계 에서 모두 필요한 확인되지 않은 mTORC1 활성과 새로운 지방 생성을 억제하여 "대사" 종양 억제 역할을 하는 것으로 나타났습니다.
우리는 최근에 세포질 분열 시작 전에 주요 표적 ACC1(FA 합성을 위한 속도 제한 효소)의 감소된 AMPK 활성화 및 인산화에 수반되는 증가된 새로운 FA 합성을 관찰했습니다.
이러한 관점에서, 새로운 FA 합성 및 막으로의 FA 혼입을 억제함으로써, AMPK의 활성화는 세포가 유사분열을 완료하는 것을 방지하여 "지방 생성" G2-M 체크포인트에서 세포를 정지시킬 것입니다.
이것은 실제로 AMPK의 직접적인 초생리학적 활성화 하에서 관찰되었습니다.
세포 주기 정지(S 단계에서 감소된 세포 분획을 통해) 및/또는 세포자멸사는 FA 합성을 직접 억제하는 ACC1 및 지방산 합성효소(FASN) siRNA를 사용하여 이전에 확인되었습니다.
AMPK는 또한 세포 주기 조절에서 직접적인 대사 독립적 역할을 합니다.
따라서 AMPK의 지속적인 초생리학적 활성화에 의해 유도된 세포 주기 정지는 새로운 FA 합성(대사 역할)과 유사분열 방추 조립/염색체 분리 이상(비대사적 역할)의 억제 모두에 기인할 수 있습니다.
최근에는 에너지 균형과 무관하게 세포 주기를 직접 조절하는 소단위 AMPK α1의 역할도 등장했습니다.
"종양 억제인자"로서 AMPK의 행동을 지지하는 세 번째 기전은 종양 유전자 BRAF의 AMPK 의존적 인산화를 보여줍니다. 이 인산화는 Ras 1(KSR1)의 스캐폴딩 단백질 키나제 억제제와의 BRAF 상호작용을 방지하여 발암성 MEK-ERK 신호 전달을 억제하고 결과적으로 세포 증식 및 세포 주기 진행의 손상을 초래합니다.
또한, 종양 성장을 억제하는 추가 작용 메커니즘이 제안되었습니다.
AMPK 활성화가 상피 세포가 암 줄기 세포와 같은 특성을 획득하고 기저막을 뚫고 먼 부위로 전이하는 능력을 얻는 것으로 생각되는 과정인 EMT를 방해한다는 것을 시사했습니다.
추가 증거는 또한 일부 종양 유형 및 유전적 맥락에서 AMPK의 종양 억제인자 역할을 뒷받침합니다.
첫째, protein kinase B(Akt)는 Ser485에서 AMPK 인산화를 유도하여 LKB1에 의한 활성화를 감소시키는 것으로 보고되었습니다
.이는 포스파타제 및 텐신 동족체(PTEN) 기능 상실 돌연변이 또는 포스포이노시티드-3-키나제(PI3K)의 활성화 돌연변이로 인해 Akt가 과활성화되는 종양에서 발생할 수 있습니다.
둘째, AMPK 활성화는 구성적으로 활성 다운스트림 ERK를 유도하는 가장 흔한 BRAF 돌연변이 V600E를 보유하는 흑색종 세포에서 억제됩니다.
셋째, AMPK 억제는 갑상선암의 PTEN 결핍 모델과 미토콘드리아 열충격 단백질 90 샤페론 TRAP-1을 발현하는 비소세포폐암(NSCLC) 세포에서 관찰되었습니다 .
넷째, 호기성 해당작용으로의 대사 전환을 특징으로 하는 유전성 평활근종증 신세포암(HLRCC) 환자의 푸마르산염 수화효소 결핍 신장 종양 및 세포주에서 AMPK 수준이 감소합니다.
AMPK 감소는 DMT1 철 수송체의 발현 감소, 세포질 철 결핍, 철 조절 단백질 IRP1 및 IRP2의 활성화로 이어져 HIF1α의 발현이 증가합니다.
HIF1α의 침묵 또는 AMPK의 활성화는 HLRCC 세포주 UOK262의 침습적 활동을 감소시키며, 이는 HIF1α의 과발현과 AMPK의 하향 조절이 푸마르산 수화효소 결핍 세포의 발암성 성장에 기여함을 나타냅니다.
종합하면, 이러한 결과는 특정 유전적, 대사적 및 신호 전달 맥락에서 AMPK가 종양 억제 역할을 할 수 있음을 시사합니다( 그림 3 ).
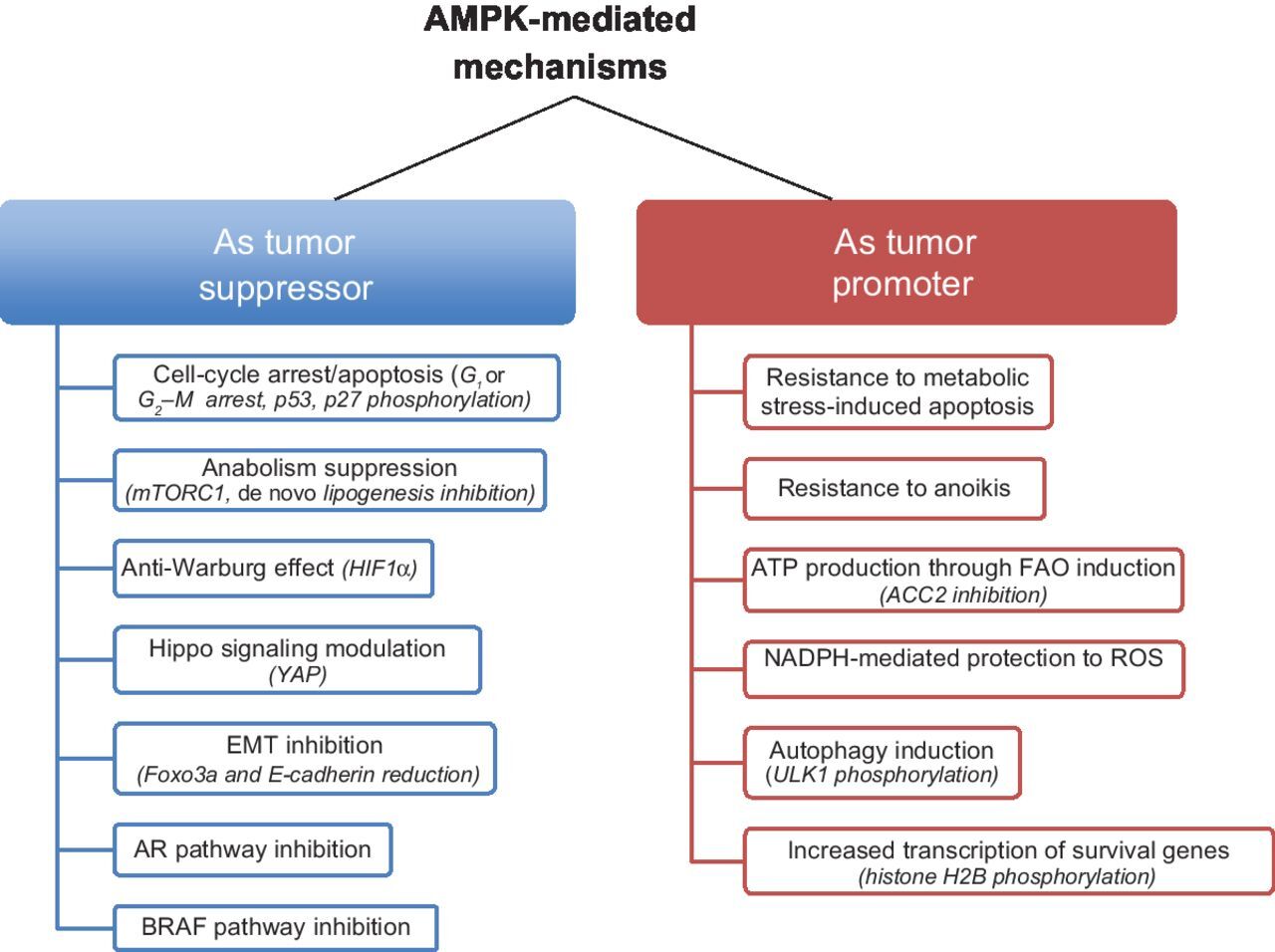
AMPK가 암에서 양면적인 역할을 할 수 있는 주요 메커니즘.
AMPK 활성화는 상황에 따라 다른 다운스트림 경로를 활성화하여 종양 발달/진행을 억제하고 촉진할 수 있는 세포 과정을 촉발합니다.
NADPH, 니코틴아미드 아데닌 디뉴클레오티드 포스페이트의 환원된 형태.
상황별 종양 촉진제로서의 AMPK
저산소증/영양소 결핍 또는 기질 분리와 같은 대사 스트레스 조건에서 생존하는 능력은 암세포의 기본입니다.
AMPK 경로가 이 가소성을 지원하는 몇 가지 메커니즘이 설명되었습니다.
여기에는
(i) unc-51-like kinases(ULK)의 AMPK 의존적 인산화에 의한 자가포식 유도,
(ii) ATP 생성을 위한 FA 산화(FAO) 촉진.
(iii) 코어 히스톤 H2B의 인산화에 의해 유도된 전사 변화 및
(iv) 세포독성 ROS를 중화하기 위한, FAO의 활성화/FA 합성의 억제를 통한 세포내 NADPH 수준의 증가( 그림 3 ).
흥미롭게도, 영양이 풍부한 조건에서 AMPK 에너지 감지 경로와 PI3K/Akt 캐스케이드는 mTOR에 의해 반대 조절 효과를 일으키지만, 포도당 고갈 상태에서는 AMPK와 Akt가 모두 활성화되어 세포 생존을 협력적으로 지원합니다.
따라서 LKB1/AMPK 경로는 종양 성장을 억제하는 능력을 통해 종양 억제인자로 작용할 수 있지만, 그것은 또한 예를 들어 종양 성장이 산소와 영양소를 전달하는 혈액 공급 용량을 초과할 때 "종양 촉진제"로 작용하여 종양 세포가 대사 스트레스에 더 저항할 수 있도록 합니다.( 그림 4 ).
AMPK 활성화는 영양이 풍부한 조건에서도 특정 종양 유형 및 유전적 맥락에서 종양 성장을 촉진할 수 있습니다.
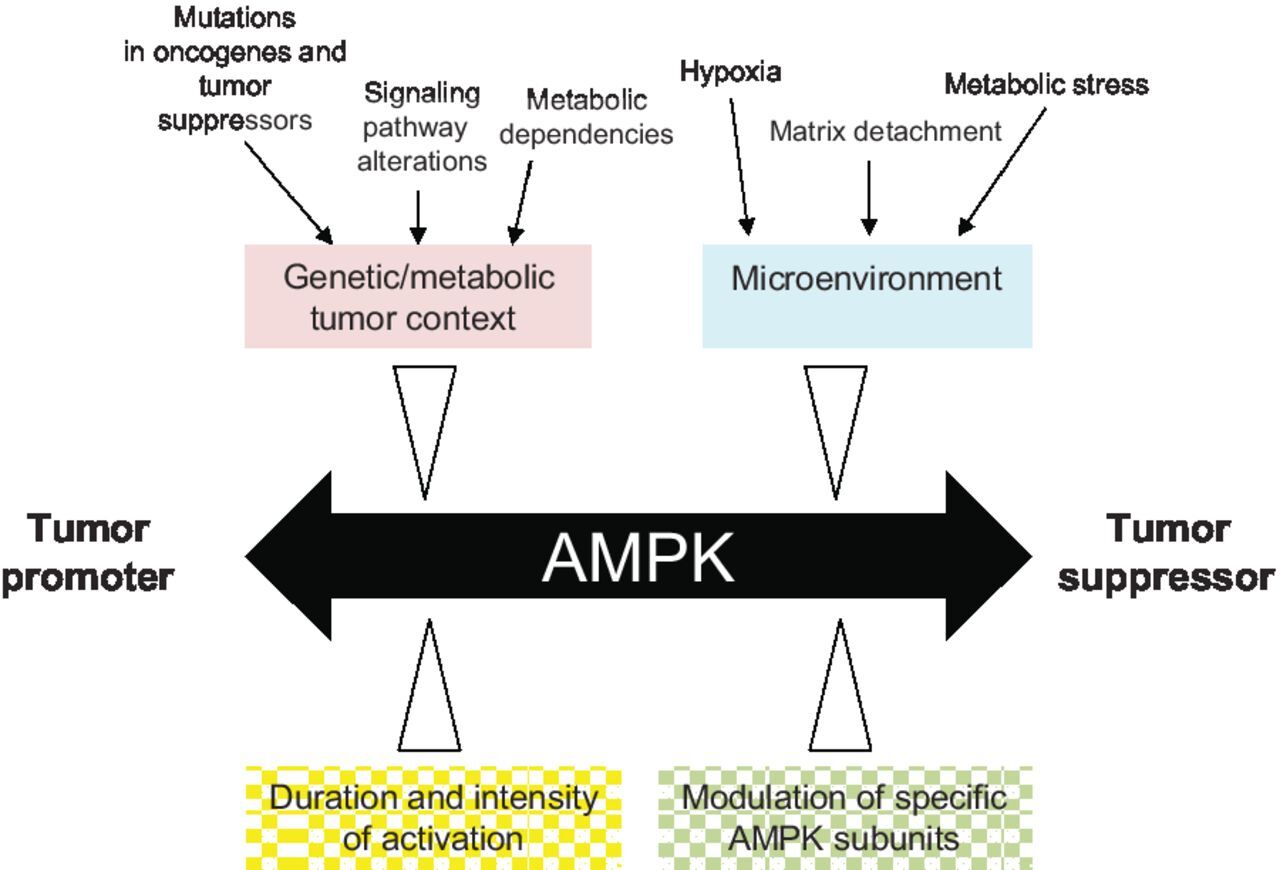
AMPK는 "조건부" 종양 억제인자 및 "상황적" 종양 촉진제로 기능합니다.
암에서 AMPK 활성화의 결과는 유전적 맥락=context, 암세포의 대사 의존성 및 주변 미세 환경의 영향을 받습니다.
이종삼량체=heterotrimer의 특정 소단위체의 발현/활성화뿐만 아니라 AMPK 활성화의 강도/지속기간(예: 생리학적 활성화 대 약물 유발 초생리학적=supraphysiological 활성화)의 차이는 다양한 암 유형에서 AMPK의 항종양형성 및 종양형성 역할에 기여합니다 .
최근 증거는 공격적인 유방 및 성상세포 종양에서 종양 성장을 지원하는 AMPK의 핵심 역할을 보여주었습니다.
AMPK 조절제의 치료 효과: 메트포르민 역설
암에서 AMPK의 이분법적 역할에 대한 더 나은 이해는 또한 암 치료에서 AMPK 조절제의 사용에 대한 신중한 재평가를 가져왔습니다.
그런 면에서 메트포르민의 경우가 상징적이다.
미토콘드리아 전자 전달 사슬의 복합체 I을 억제하여 세포 내 ADP, AMP 및 에너지 스트레스 수준을 증가시키는 비구아니드 메트포르민 및 펜포르민뿐만 아니라 천연 화합물인 AMP 모방 약물 AICAR과 함께 달성되었습니다(참고 문헌에서 검토됨).
그러나 메트포르민의 생체 내 항종양 특성 을 AMPK 활성화로 돌리는 것은 약물의 주요 효과가 간의 글루코스 신생합성을 억제하여 종양 세포 증식의 잘 알려진 두 가지 촉진제인 포도당과 인슐린의 순환 수준을 감소시키기 때문에 비판을 받아왔습니다.
이것은 여러 AMPK와 무관한 메커니즘이 설명 된 in vitro에서 메트포르민의 항종양 효과에도 유효합니다.
더욱이, 소위 "비구아나이드 역설"의 발견은 최근 특정 상황에서 메트포르민 매개 종양 성장 억제가 AMPK 활성화에 의존하지 않고 오히려 하향 조절에 의존한다는 것을 시사했습니다.
LKB1/AMPK 경로에 결함이 있는 세포는 메트포르민 치료로 유발된 대사 스트레스에 반응하여 ATP 수준을 회복할 수 없기 때문에 LKB1/AMPK가 결핍된 암세포는 기능적 LKB1/AMPK 축을 가진 세포보다 세포 사멸에 더 취약합니다. 그림 5 ).
이에 비추어 볼 때, 비구아니드의 사용은 AMPK를 활성화하기보다는 억제하는 제제와 함께 사용하는 것이 가장 효과적일 수 있으며, 전반적으로 이러한 데이터는 활성제보다 AMPK 억제제를 사용하는 것이 맥락에서 암 세포 사멸을 우선적으로 촉발할 것임을 시사합니다.
대사 스트레스. 흥미롭게도 화학요법제인 수니티닙은 AMPK를 억제하는 것으로 나타났으며, 이는 수니티닙과 메트포르민의 병용 치료가 임상적으로 관련이 있을 수 있음을 시사합니다.
그림.5
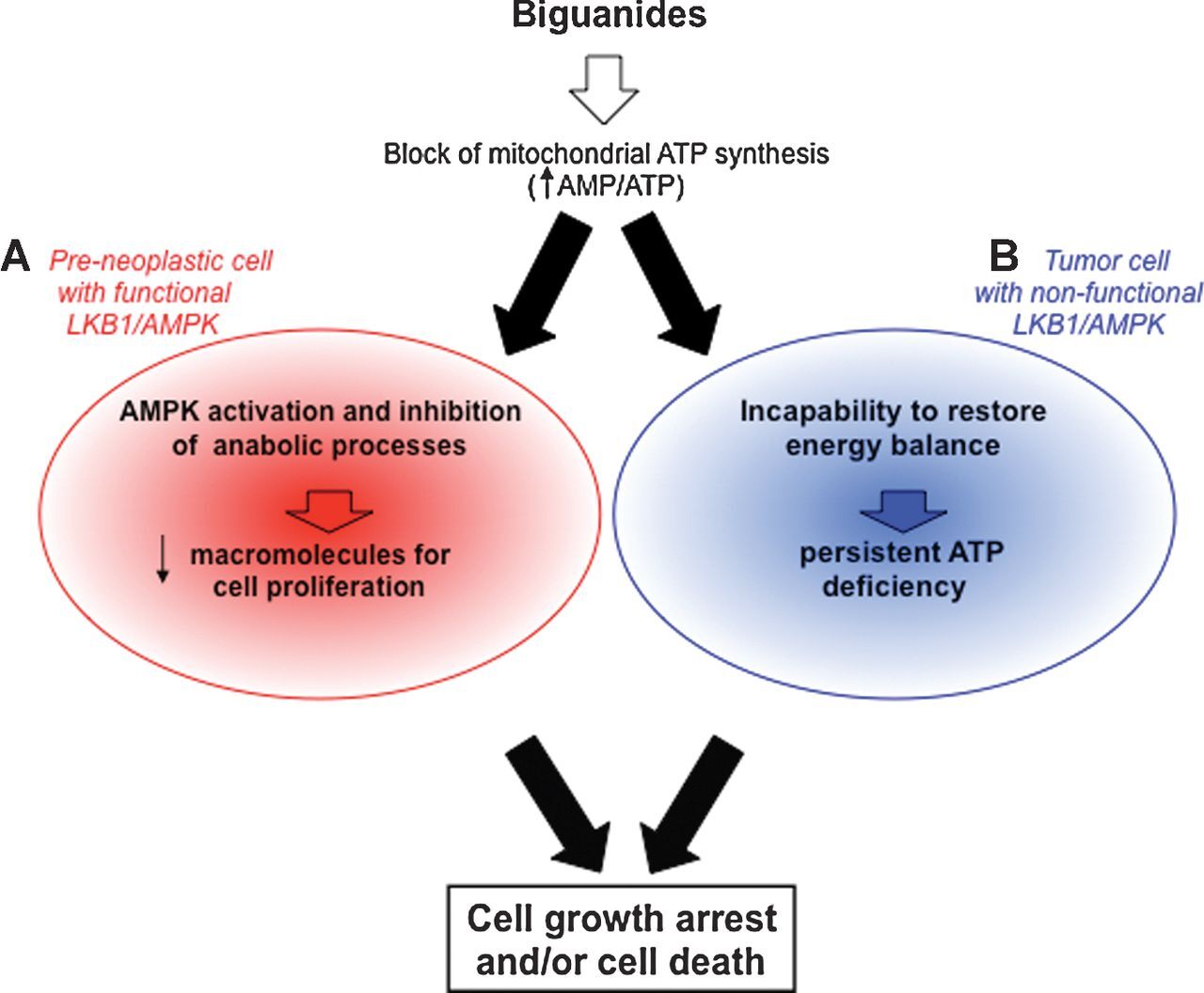
비구아니드가 LKB1 양성 및 음성 종양에서 치료학적으로 유익한 기전.
A, 메트포르민 또는 펜포르민은 기능적 LKB1/AMPK 경로로 전암 세포에서 AMPK를 활성화하여 성장 및 증식을 억제하여 종양 형성의 시작을 지연시킵니다.
B, LKB1-AMPK 경로가 기능하지 않는 암세포는 비구아나이드로 인한 에너지 스트레스를 회복할 수 없으며 세포 사멸에 더 민감합니다(비구아니드 역설).
메트포르민과 AICAR 치료의 표적 외 효과를 극복하기 위해 새로운 직접 AMPK 활성제가 개발되었습니다.
OSU-53에 의한 AMPK 활성화가 Foxo3a를 활성화하여 유방암 및 전립선암 세포에서 "EMT"를 차단하여 시험관 내 침습성 표현형 과 생체 내 전이 특성 을 억제한다고 보고했습니다.
영양이 풍부한 조건에서 AMPK의 직접적인 초생리학적 활성화는 유사분열 정지 및 세포자살과 관련하여 전립선암 세포의 성장을 억제하고 시험관 내에서 항안드로겐의 효과를 강화하는 것으로 나타났습니다
마지막으로, 새로운 AMPK 활성화제인 화합물 1 은 시험관 내에서 상당한 항종양 활성을 유도하고 결장직장암의 마우스 이종이식 모델에서 종양 성장 지연을 유도합니다.
그러나 화합물 1이 AMPK를 활성화하는 메커니즘은 아직 밝혀지지 않았습니다.
종합하면, AMPK의 지속적인 초생리학적 활성화의 유도는 일부 암 유형에서 종양 억제를 초래합니다( 그림 3 ).
장에서 흡수된 후 아스피린의 활성 대사산물인 살리실산염은 최근 A-769662와 같은 β1 소단위체의 동일한 부위에 결합하는 직접적인 AMPK 활성제로 확인되었습니다.
이것은 AMPK 활성화가 암에 대한 아스피린의 보호 효과를 매개하는데 관여할 수 있음을 시사합니다.
그러나 이 가설을 검증하기 위해서는 유전적으로 조작된 AMPK 모델에 대한 향후 전임상 연구가 필요합니다.
전반적으로, 이러한 명백히 상충되는 데이터는 AMPK 활성화제와 억제제가 모두 다양한 종양 유형, 다양한 유전적/대사적 맥락 및 다양한 미세환경 조건에서 치료적 이점을 제공할 수 있음을 시사합니다.
따라서, AMPK 조절제의 선택은 종양 형성/종양 진행의 다양한 단계에서 다를 수 있습니다.
암에서의 AMPK 역할: 인간 연구
인간 암에서의 AMPK 활성화
인간 조직에서 AMPK 활성화의 평가는 간단하지 않습니다.
초기 연구에서는 동결 클램핑이 아닌 주변 온도에서 해부로 조직과 장기를 제거할 때 ACC 인산화가 사후 인공물로 발생했음을 보여주었습니다.
상온에서의 절개는 아마도 혈액 공급 중단에 따른 저산소증으로 인한 AMP 상승 및 ATP 고갈로 이어져 AMPK 활성화를 초래합니다.
더욱이, 간과 같은 조직에서 ACC 인산화는 주간 리듬을 따르고 식이 행동의 영향을 받는 것으로 나타났습니다.
따라서 인간 조직에서 AMPK 활성 및 ACC 인산화 분석은 신중하게 해석되어야 합니다.
전반적으로, 이러한 인간 연구는 AMPK 활성화가 여러 암 유형에서 질병 진행을 지연시킬 수 있다는 가설을 뒷받침합니다.
1 번 테이블.
종양 조직에서 AMPK 활성화, 임상 병리학 적 특징 및 예후에 대한 인구 기반 연구
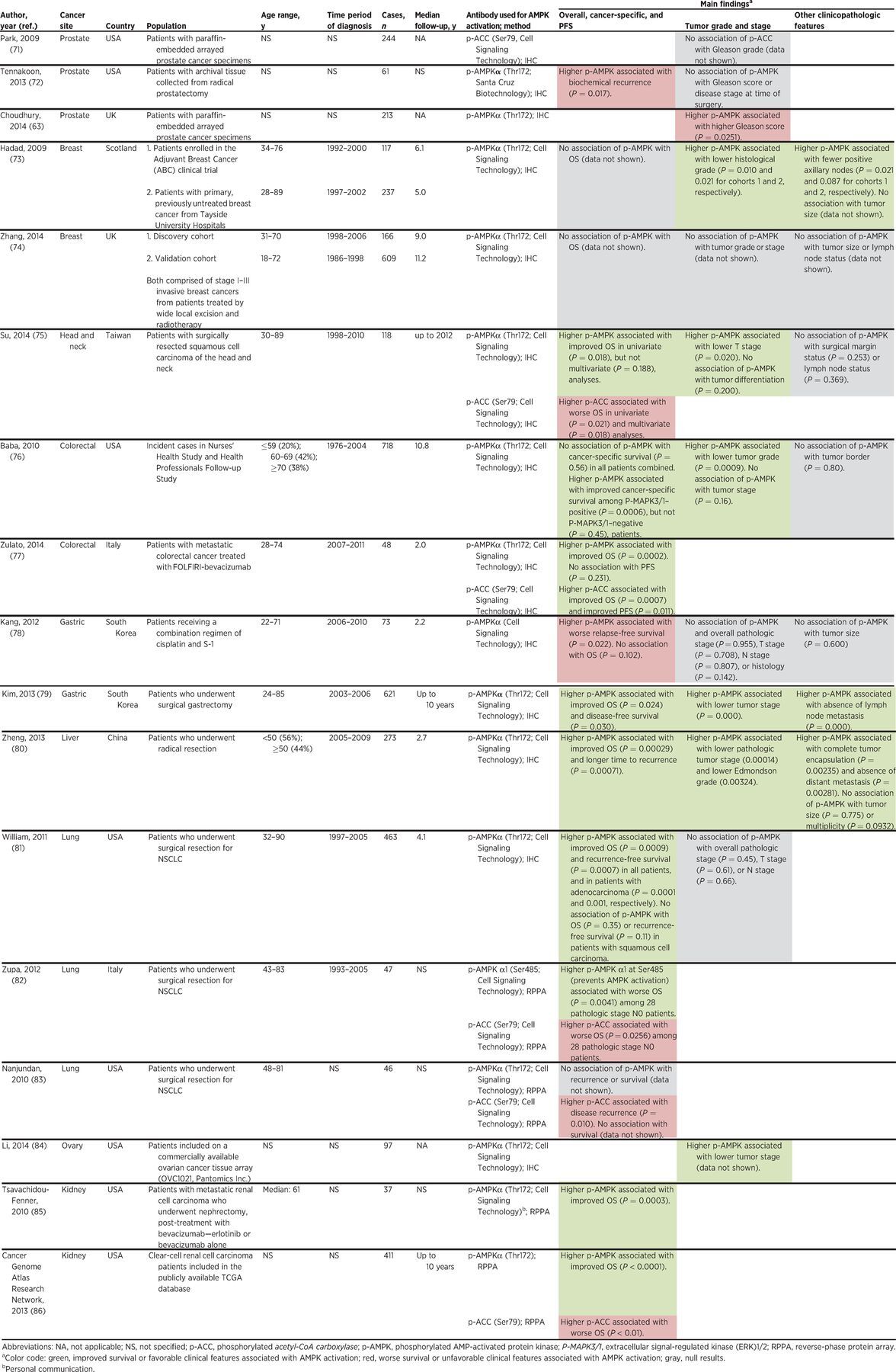
요약하면, AMPK 발현/활성화는 종양 단계 및 조직학, 임상 결과 및 조직 유형(정상, 종양 및 전이)에 따라 다릅니다.
종양 조직에 대한 대부분의 연구는 Thr172에서의 인산화에 의해 측정된 AMPK 활성화가 종양 진행을 지연시키는 역할을 지지합니다.
그러나 종양을 비종양 조직과 비교하면 AMPK가 종양 개시에 관여할 수 있음을 시사합니다.
따라서 인간 연구의 증거는 또한 발암에서 AMPK의 이중 역할을 강조합니다.
인간의 AMPK 활성화 약물: 메트포르민, 펜포르민 및 아스피린
전반적으로, 문헌은 메트포르민이 암 위험을 감소시키거나 전혀 영향을 미치지 않는다고 제안하지만, 비당뇨병 인구에서 메트포르민 사용을 다룬 연구는 거의 없습니다.
일반 인구를 대상으로 한 메트포르민 요법의 향후 임상 시험은 메트포르민을 화학 예방제로 사용할 가능성에 대한 중요한 데이터를 제공해야 합니다.
메트포르민 사용은 또한 암 진단 후 질병 진행에 영향을 미칠 수 있습니다.
관찰 연구에서 메트포르민은 전립선암, 다발성 골수종, 간암, 난소/난소 암, 자궁내막암, 방광암, 유방암 환자 집단에서 질병 재발, 전체 사망률 또는 암 관련 사망률 감소 위험과 관련이 있었습니다.
근치적 전립선 절제술을 받은 전립선암 환자에 대한 2건의 추가 연구에서 메트포르민 사용과 생화학적 재발까지의 시간 또는 장기적인 결과 사이에 유의미한 연관성이 발견되지 않았습니다.
유방암 환자에 대한 2개의 추가 연구는 메트포르민 사용 및 전체 또는 암 특이적 생존에 대해 무효였습니다
단기 메트포르민 사용과 표적 조직에서 AMPK 활성화 사이의 직접적인 연관성은 불분명합니다.
메트포르민이 AMPK 경로를 통해 종양 성장 및 진행에 영향을 미치는지 여부를 결정하려면 다양한 암 유형에 걸쳐 메트포르민 사용의 더 긴 기간과 다양한 용량에 대한 대규모 연구가 필요합니다.
연구자들은 모든 비구아나이드를 그룹으로 분석한 결과 비당뇨병에 비해 비구아니드 사용자의 대장암 위험이 증가한다는 사실을 발견했으며, 이러한 결과는 비구아나이드 치료와 결장직장암 발병률이 감소하거나 연관성이 없음을 시사하는 현재 문헌의 많은 부분과 상충됩니다.
보다 최근에는 아스피린의 대사 유도체인 살리실산이 AMPK를 직접 활성화하는 것으로 나타났습니다
아스피린은 항종양 성질을 나타내는 것으로 오랫동안 알려져 왔지만 이러한 성질이 AMPK에 의해 매개되는지 여부는 알려져 있지 않습니다.
추가적인 관찰 연구는 진단 후 정기적인 아스피린 사용과 유방, 결장직장 및 전립선암 환자 사이의 개선된 생존 결과 사이의 연관성을 지지하는 반면, 다른 연구에서는 그렇지 않습니다.
전반적으로, 장기간의 관찰 및 무작위 연구의 현재 증거는 암의 1차 및 2차 예방에서 아스피린의 잠재적 역할을 강력하게 시사합니다.
요약하면, 관찰 및 무작위 연구는 화학 예방 및/또는 암 생존 개선을 위한 AMPK 활성화 약물의 잠재적인 이점을 시사합니다.
이러한 발견은 종양 조직의 AMPK 활성화 수준과 여러 암 유형에서 관찰된 보다 유리한 임상병리학적 특징 및 생존 결과 사이의 연관성과 일치합니다( 표 1 ).
결론
암세포에서 AMPK 활성화의 이중 역할은 상황에 따라 다르며 AMPK 조절 결과에 영향을 미칩니다.
********************************************************************************
Targeting AMPK for cancer prevention and treatment
AMP-activated protein kinase (AMPK) is an important mediator in maintaining cellular energy homeostasis. AMPK is activated in response to a shortage of energy. Once activated, AMPK can promote ATP production and regulate metabolic energy. AMPK is a known .
www.ncbi.nlm.nih.gov
암 예방 및 치료를 위한 AMPK 표적화
메트포르민
메트포르민의 이점을 위해 AMPK 활성화가 필요한지 여부에 대한 불확실성이 남아 있지만 메트포르민이 AMPK를 활성화할 수 있고 암 예방 및 치료에 유용할 수 있다는 것은 여전히 분명합니다.
비스테로이드성 소염제(NSAID)
염증은 종양의 시작, 진행 및 전이에 중요한 역할을 하는 것으로 나타났습니다.
비스테로이드성 소염제(NSAID)인 아스피린은 특히 대장암(CRC)을 예방하는 암 발병 위험 감소와 상관관계가 있는 것으로 나타났습니다
흥미롭게도 아스피린과 다른 NSAIDs도 AMPK를 활성화시키는 것으로 나타났습니다.
결장직장암 세포를 아스피린으로 치료하면 AMPK 활성화가 크게 증가하고 하류 mTOR 신호 전달이 억제되었습니다
아스피린이나 다른 NSAID가 AMPK를 활성화하면 다른 염증 조절 경로가 조절될 가능성이 있습니다.
예를 들어 폐에서 AMPK 활성화는 대식세포, 호중구 및 T 세포의 활성을 직접 조절하여 손상이나 감염으로 인한 염증을 완화할 수 있습니다.
백혈구와 그 사이토카인은 종양 발달의 모든 측면에서 중요한 조절 역할을 합니다.
따라서 AMPK 활성화를 표적으로 하는 항염증제는 암 관련 염증 치료에 더 의미 있는 역할을 할 수 있습니다.
천연 제품
관찰 연구에 따르면 일부 천연물이 암 발병을 예방하는 효능이 있습니다.
그러나 대부분의 경우 이 개념을 뒷받침하는 임상 시험이 아직 수행되지 않았습니다.
(그림(그림11및 테이블1 번 테이블).1).
wogonin, tanshinone IIA , quercetin 및 cryptotanshinone 과 같은 플라본은 다양한 유형의 암세포에서 증식을 억제하고 세포 사멸을 유도하는 AMPK 활성화를 유도합니다.
폴리페놀은 AMPK 활성화제의 풍부한 공급원이기도 합니다.
레스베라트롤은 AMPK/mTOR 경로를 조절하여 만성 골수성 백혈병 세포에서 자가포식을 유도합니다.
결장직장암 세포에서 magnolol, epigallocatechin-3-gallate (EGCG) 및 widdrol 은 세포 사멸을 유도하고 이동을 억제하며 AMPK 의존성 메커니즘에 의한 침입을 방지할 수 있습니다.
유방암 세포에서 Nordihydroguaiaretic acid는 mTOR-Raptor 복합체의 파괴와 AMPK 활성화를 통해 mTORC1 활성을 억제하는 반면, 강력한 AMPK 활성화제인 demethoxycurcumin 은 모 화합물과 함께 삼중 음성 유방암 세포에서 광범위한 항암 활성을 나타냅니다
커큐민은 AMPK-p53 활성화에 의해 난소암 세포와 결장직장암 세포 에서 유사한 효과를 발휘 합니다.
Antrodiacamphorate에서 추출한 Antroquinonol 은 AMPK 의존적으로 간세포암종 세포에 대한 항암 활성을 나타내며, 또한 honokiol 역시 AMPK 의존 메커니즘을 통해 여러 암세포 유형에서 증식을 억제하는 것으로 나타났습니다.
Berberine은 AMPK의 활성화를 통해 AOM/DSS 마우스 모델에서 결장 종양 형성을 억제하는 것으로 나타났습니다.
Berberine 또는 Ginsenoside 20-ObD-Glucopyranosyl-20(S)-Protopanaxadiol 은 시험관 내에서 AMPK의 활성화를 통해 흑색종 세포 성장과 침입을 억제합니다.
따라서 이러한 제제는 AMPK 활성화제 역할을 할 수 있으며 천연물과 암 예방 및 치료 사이에 중요한 연결 고리를 제공할 수 있습니다.
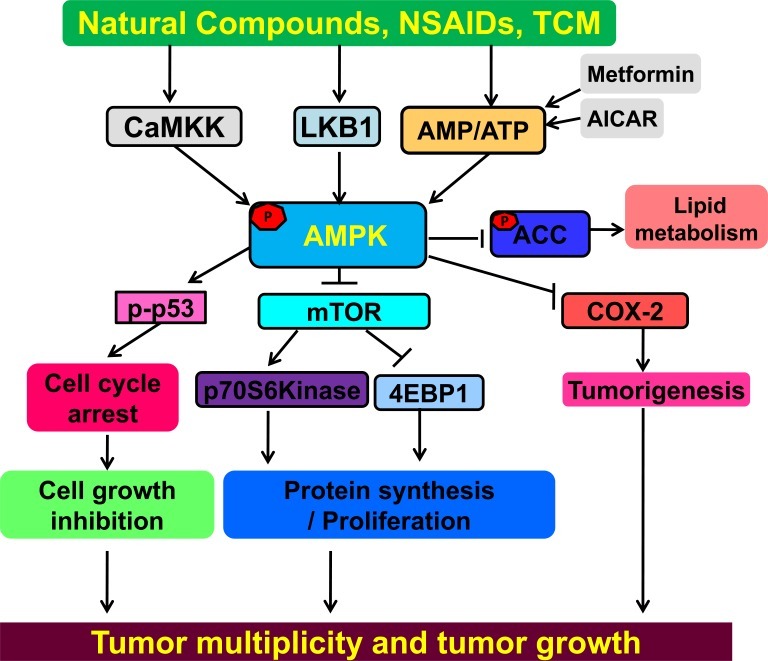
AMPK는 암 예방 및 치료를 위한 종양 억제제입니다.
NSAID, 천연 제품, TCM 및 메트포르민은 모두 AMPK를 활성화할 수 있습니다.
AMPK는 mTOR 신호 경로를 부정적으로 조절하여 암 증식과 성장을 억제합니다.
활성화된 AMPK는 종양 형성과 관련된 염증 유발 효소인 COX-2를 부정적으로 조절합니다.
AMPK는 종양 억제 인자 p53의 인산화를 유도하여 세포 주기를 정지시킬 수 있습니다.
AMPK의 활성화는 또한 지질 대사에 영향을 미치는 ACC의 인산화를 유도할 수 있습니다.
분자 표적의 활성화로 이어지는 상호 작용은 화살표로 표시됩니다.
억제된 것은 막대로 표시됩니다.
AMPK의 활성화는 항암 활동으로 이어지는 여러 경로를 조절할 수 있습니다.
TCM=중국 전통 의학; NSAIDs=비스테로이드성 항염증제.
1 번 테이블
| 베르베린 | AMPK/ERK에 의한 억제 전이 | 흑색종 | |
| 진세노사이드 20-ObD-글루코피라노실 -20(S)-프로토파낙사디올 |
AMPK/JNK에 의한 자가포식 세포사 유도 | 흑색종 | |
| Wogonin | AMPK/mTOR/4EBP1에 의한 억제 번역 | 교모세포종 | |
| Tanshinone IIA | AMPK/mTOR/p70S6kinase에 의한 자가포식 세포사 유도 | 백혈병 | |
| 케르세틴 | AMPK/COX-2를 통한 성장 억제 | 유방암 및 결장암 | |
| Cryptotanshinone | AMPK/mTOR에 의한 자가포식 세포사 유도 | 간암 및 결장암 | |
| 레스베라트롤 | AMPK/mTORC2/p62를 통한 자가포식 세포 사멸 유발 | 백혈병 | |
| Magnolol | AMPK/p53에 의한 결장암의 세포자살 유도 | 대장 암 | |
| 에피갈로카테킨-3-갈레이트 | ROS/AMPK/COX-2에 의한 대장암 증식 억제 | 대장 암 | |
| Widdrol | AMPK를 통한 세포자멸사 유도 | 대장 암 | |
| Nordihydroguaiaretic acid | AMPK/mTORC1에 의한 유방암 성장 억제 | 유방암 | |
| Demethoxycurcumin | AMPK/mTORC1에 의한 유방 세포 성장 억제 | 유방암 | |
| 커큐민 | AMPK/p53에 의한 증식 억제 | 난소 암 | |
| Antroquinonol | AMPK/mTOR/p70s6kinase 및 4EBP1에 의한 항암 | 간세포 암 | |
| 호노키올 | LKB1/AMPK/mTOR에 의한 유방암의 침윤 및 이동 억제 | 유방암 | |
| 베르베린 | AMPK/mTOR 및 AMPK/COX-2에 의한 억제 성장 | 대장 암 |
AICAR
5-aminoimidazole-4-carboxamide ribonucleotide(AICAR)는 AMPK의 약리학적 활성제입니다.
중국 전통 의술(Traditional Chinese medicine=TCM)
TCM 제제에 의한 AMPK 활성화는 메트포르민에서 볼 수 있는 유사한 결과를 생성했습니다.
따라서 중국 전통 의학에서 만성 염증과 당뇨병을 치료하는 데 사용되는 많은 약초는 암 예방 및 치료를 위해 AMPK를 표적으로 삼는 잠재적인 후보입니다.
AMPK 활성화의 잠재적인 단점
AMPK를 표적으로 하는 것이 암 치료의 매력적인 표적이 되었지만 AMPK 활성화가 암을 촉진할 수 있는 경우가 있습니다.
mTOR의 경우 isoform 특이성이 특히 중요합니다.
대부분의 암은 위에서 논의한 바와 같이 4EBP1 및 S6K1과 같은 이펙터를 통해 성장을 조절하는 mTOR 복합체 1(mTORC1)에서 활성화됩니다.
따라서 mTORC1을 억제하면 세포 단백질 합성 및 증식이 방지됩니다.
그러나 mTOR 복합체 2(mTORC2)를 억제하지 않고 mTORC1을 억제하면 앞서 언급한 바와 같이 PI3K-Akt 신호 전달 경로를 활성화하고 종양 생존을 촉진할 수 있습니다.
대부분의 경우 AMPK가 활성화되면 mTORC1이 억제됩니다.
그러나 mTORC2 및 Akt 활성화에 대한 효과는 불완전하게 이해됩니다.
특정 AMPK 활성제가 mTOR를 조절하기 위해서만 사용된다면 그 성공은 두 복합체를 모두 억제하는 능력에 달려 있습니다.
최근 연구에서 AMPK 작용제는 종양 억제 및 종양 촉진 능력을 모두 가지고 있는 것으로 나타났으며, 이는 피드백 조절과 관련된 메커니즘에서 기인할 수 있습니다..
전립선암의 경우 AMPK 활성화는 불량한 예후와 관련될 수 있습니다.
암 치료의 경우 AMPK 활성화는 세포 유형 및 상황에 따라 달라질 수 있으며 향후 연구에서 해결하기 가장 어려운 난제 중 하나가 될 것입니다.
미래 전망
상당한 양의 증거가 AMPK 활성화가 대사성 종양 억제인자로 작용할 수 있다는 개념을 뒷받침합니다.
AMPK 활성화는 암이 발달하고 진행하는 데 필요한 기본 요구 사항 중 하나를 표적으로 하는 세포 대사를 재프로그래밍함으로써 종양 발달에 반대할 수 있다고 가정합니다.
AMPK가 NSAIDs와 전통적으로 항염증제로 간주되는 약제에 의해 활성화된다는 점을 고려하면 AMPK 활성화의 화학 예방 활성이 염증 조절 능력과 관련이 있는지 여부에 대한 중요한 질문이 제기됩니다.
현재 AMPK와 관련된 대부분의 연구는 신진대사와 관련되어 있으며 최근에야 염증 과정에서 AMPK의 직접적인 역할과 이것이 메트포르민, NSAID, TCM 및 기타 AMPK 활성제에서 볼 수 있는 항암 활성과 어떻게 관련될 수 있는지를 밝히기 시작했습니다.
또한 아직 조사해야 하는 AMPK 활성화의 다른 모든 간접 효과가 있습니다.
따라서 AMPK 활성제가 암 예방 및 치료에 임상적으로 사용되기 위해서는 추가적인 연구가 필요하다.
**********************************************************************************
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4480686/
Targeting AMPK for cancer prevention and treatment
AMP-activated protein kinase (AMPK) is an important mediator in maintaining cellular energy homeostasis. AMPK is activated in response to a shortage of energy. Once activated, AMPK can promote ATP production and regulate metabolic energy. AMPK is a known .
www.ncbi.nlm.nih.gov
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5789788/
The double-edged sword of AMPK signaling in cancer and its therapeutic implications
5′-AMP-activated protein kinase (AMPK) plays a pivotal role in maintaining energy and redox homeostasis under various metabolic stress conditions. Metabolic adaptation, which can be triggered by the activation of AMPK during metabolic stress, ...
www.ncbi.nlm.nih.gov
https://bmccancer.biomedcentral.com/articles/10.1186/s12885-022-09211-1
The mixed blessing of AMPK signaling in Cancer treatments - BMC Cancer
Background Nutrient acquisition and metabolism pathways are altered in cancer cells to meet bioenergetic and biosynthetic demands. A major regulator of cellular metabolism and energy homeostasis, in normal and cancer cells, is AMP-activated protein kinase
bmccancer.biomedcentral.com
https://royalsocietypublishing.org/doi/10.1098/rsob.190099
The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde?† | Open Biology
The AMP-activated protein kinase (AMPK) acts as a cellular energy sensor. Once switched on by increases in cellular AMP : ATP ratios, it acts to restore energy homeostasis by switching on catabolic pathways while switching off cell growth and ...
royalsocietypublishing.org
https://www.sciencedirect.com/science/article/abs/pii/S1044579X21001073
AMPK signaling and its targeting in cancer progression and treatment
The intrinsic mechanisms sensing the imbalance of energy in cells are pivotal for cell survival under various environmental insults. AMP-activated pro…
www.sciencedirect.com
'암치료' 카테고리의 다른 글
| 암의 지질 대사 환경과 새로운 치료적 관점 (0) | 2022.09.27 |
|---|---|
| 암 진행 및 치료 전략에서의 지질 대사 (프로토콜) (0) | 2022.09.26 |
| (링크) Full Metabolic Blockage(좋은습관님) (0) | 2022.08.11 |
| (링크)APOTOSIS 세포자멸사, 암 대사 치료의 종착역(좋은습관님) (0) | 2022.08.11 |
| (링크)오프라벨 드럭 부작용 및 상호작용 정리(좋은습관님) (0) | 2022.08.11 |