https://www.annualreviews.org/doi/full/10.1146/annurev-cancerbio-041916-065808
The Two Faces of Reactive Oxygen Species in Cancer
Reactive oxygen species (ROS), now appreciated for their cellular signaling capabilities, have a dual role in cancer. On the one hand, ROS can promote protumorigenic signaling, facilitating cancer cell proliferation, survival, and adaptation to hypoxia. On
www.annualreviews.org
The Two Faces of Reactive Oxygen Species in Cancer
2017
현재 세포 신호 전달 능력으로 인정받고 있는 활성산소종(ROS)은 암에서 이중 역할을 합니다.
한편으로, ROS는 암 세포 증식, 생존 및 저산소증에 대한 적응을 촉진하는 protumorigenic 신호를 촉진할 수 있습니다.
반면에, ROS는 항종양 신호를 촉진하고 산화 스트레스로 인한 암세포 사멸을 유발할 수 있습니다.
세포 변형 및 종양 형성에 필요한 세포 신호 전달 경로를 과활성화하기 위해 암세포는 정상 세포에 비해 ROS 생성 속도를 증가시킵니다.
동시에 ROS 항상성을 유지하고 세포 사멸을 피하기 위해 암세포는 항산화 능력을 증가시킵니다.
정상 세포와 비교하여 암세포의 이러한 변경된 산화환원 환경은 ROS 조작 요법에 대한 감수성을 증가시킬 수 있습니다
암세포에 대한 초기 분석은 형질전환되지 않은 정상 세포에 비해 ROS 수준이 높은 것으로 나타났습니다.
높은 수준의 ROS는 지질, 단백질 및 DNA에 손상을 줄 수 있기 때문에 ROS는 발암성이며 게놈 불안정성과 종양 형성을 촉진하는 것으로 생각되었습니다.
ROS가 불안정한 게놈에 기여할 수 있지만 암 세포 증식, 생존, 혈관 신생 및 전이를 촉진하는 신호 분자로서의 ROS의 역할이 등장했습니다.
protumorigenic 신호 이벤트를 활성화하기 위해 암세포는 발암 돌연변이를 획득하고 종양 억제 인자를 잃고 대사를 증가시키고 저산소증 (즉, 낮은 산소 수준)에 적응함으로써 ROS 생성 속도를 증가시킵니다.
그러나 암에서 ROS의 역할은 일방적인 것이 아닙니다. 과도한 수준의 ROS는 산화 스트레스를 증가시키고 암세포 사멸을 유발할 수 있습니다.
ROS의 축적을 방지하고 산화 환원 균형을 유지하기 위해 암세포는 항산화 능력을 증가시킵니다.
과량의 ROS를 제거함으로써 암세포는 암세포 사멸을 유도하지 않고 protumorigenic 신호 경로의 활성화를 허용하는 수준에서 ROS를 유지합니다.
따라서 정상 세포와 비교하여 암세포는 높은 ROS 소거 속도에 의해 균형을 이루는 높은 ROS 생산 속도와 함께 변경된 산화환원 환경을 가지고 있습니다.
암세포의 이러한 독특한 특성은 정상 세포보다 ROS 수준의 변화나 산화환원 조작에 더 민감하게 만들 수 있습니다.
따라서 ROS를 제거하거나 ROS를 생성하는 전략은 잠재적으로 효과적인 암 치료법이 될 수 있습니다.
이 검토는 ROS의 pro- 및 antitumorigenic 신호 전달 기능과 이들의 조작이 암 치료에 어떻게 사용될 수 있는지에 중점을 둡니다.
ROS 생성 및 규제
ROS의 종류
ROS는 슈퍼옥사이드 음이온(O 2 - ), 과산화수소(H 2 O 2 ) 및 하이드록실 라디칼(OH • ) 을 포함하는 산소 함유 분자입니다 .
이 종은 오랫동안 암과 같은 병리를 유발하는 독점적인 독성 분자로 여겨져 왔습니다.
그러나 이제 ROS는 성장 인자 신호 전달, 증식 및 저산소증에 대한 적응을 포함하여 수많은 생리학적 및 생물학적 반응을 조절하는 신호 분자 역할을 할 수 있음이 인정됩니다.
H 2 O 2 는 가장 잘 설명된 신호 분자이며, H 2 O 2가 신호 전달을 매개 하는 메커니즘 은 단백질 내의 시스테인 잔기를 산화시켜 기능을 수정하는 것입니다.
H 2 O 2 는 각각 세포질, 미토콘드리아 기질 및 세포외 공간에 위치한 슈퍼옥사이드 디스뮤타제 1, 2, 3(SOD1, 2, 3)의 효소 활동성에 의한 O 2 - 의 급속한 변화에서 파생됩니다. .
O 2 - 는 또한 산화환원 신호 전달과 세포 사멸의 시작에서 기능하는 것으로 나타났습니다
OH • , H 2 O 2 가 철 또는 제 1 구리 이온과 Fenton 화학 반응을 겪을 때 형성 되며 지질, 단백질 및 DNA를 무차별적으로 손상시킵니다.
ROS의 소스
신호 전달과 관련된 ROS는 미토콘드리아와 막 결합 NADPH 산화효소(NOX)의 두 가지 주요 공급원에서 생성됩니다.그림 1)
정상적인 세포 호흡 동안 전자는 일련의 미토콘드리아 복합체를 통해 말단 전자 수용체인 분자 산소(O 2 )로 전달됩니다.
이 과정에서 전자가 전자 수송 사슬에서 누출되어 O 2 와 반응 하여 O 2 - 를 생성할 수 있습니다.
미토콘드리아에서 O 2 - 생산 의 10개 부위 중 미토콘드리아 복합체 I, II, III에서 생성된 O 2 - 는 산화환원 신호 전달에 역할을 하는 것으로 나타났습니다.
O2-는 복합체 I, II 및 III에 의해 미토콘드리아 기질로 방출되며, 여기서 SOD2는 이를 빠르게 H2O2로 전환합니다.
또한, 복합체 III는 O 2 - 를 막간 공간으로 방출 하여 O 2 - 가 전압 의존성 음이온 채널을 통해 세포질로 이동하고 SOD1에 의해 H 2 O 2 로 전환되도록 할 수 있습니다
SOD1은 또한 미토콘드리아 막간 공간에서 O 2 - 를 해독 하여 자유롭게 확산 가능한 H 2 O 2 를 생성할 수 있습니다 .

그림 1 활성산소(ROS) 생성 및 조절.
막 결합 NADPH 산화효소(NOX)와 미토콘드리아는 ROS 생산의 두 가지 주요 공급원입니다.
세포질 슈퍼옥사이드 디스뮤타제 1(SOD1)은 NOX 및 미토콘드리아 유래 슈퍼옥사이드(O 2 - )를 과산화수소(H 2 O 2 )로 전환합니다.
미토콘드리아 전자 수송 사슬(ETC)에 의해 생성된 O 2 - 는 또한 미토콘드리아 기질로 방출될 수 있고 슈퍼옥사이드 디스뮤타제 2(SOD2)에 의해 H 2 O 2 로 전환될 수 있습니다 .
H2O2는 미토콘드리아 막을 가로질러 자유롭게 확산되거나 퍼옥시레독신(PRX) 및 글루타티온 퍼옥시다제(GPX)에 의해 미토콘드리아에서 물(H2O)로 해독될 수 있습니다.
세포질에서 H2O2는 금속 양이온(Fe2+ 또는 Cu+)과 반응하여 DNA, 지질 및 단백질을 무차별적으로 손상시키는 하이드록실 라디칼(OH•)을 생성할 수 있습니다.
이는 PRX, GPX 및 카탈라아제(CAT)에 의해 H 2 O 로 해독되거나 또는 단백질 티올 산화를 통해 세포 신호를 제어합니다.
H 2 O 2 수준이 너무 낮으면 세포 신호 전달이 중단되어 항상성의 손실을 초래할 수 있습니다.
그러나 H 2 O 2 수준이 너무 높으면 비정상적인 세포 신호 전달이 발생하여 암과 같은 병리를 유발할 수 있습니다. 통제되지 않은 H2 O 2 수준은 산화 스트레스와 세포 사멸을 유발할 수 있습니다.
따라서 세포 항상성을 위해서는 최적의 H 2 O 2 수준이 필요합니다.
NOX 는 O 2 와 NADPH 로부터 세포내 및 세포외 O 2 - 생성을 촉매합니다 .
주로 원형질막에 국한되어 있지만 NOX는 핵, 미토콘드리아, 소포체를 포함한 다른 막에서도 발견될 수 있습니다.
SOD1과 SOD3는 NOX에서 파생된 O 2 - 를 H 2 O 2 로 빠르게 전환시키며 , 이는 세포막을 가로질러 쉽게 확산되어 세포 신호에 영향을 줄 수 있습니다.
중요하게도, 미토콘드리아와 NOXs는 산화제 수용체에 공간적으로 국소화하여 ROS 의존적 신호를 중재할 수 있다
그러나 ROS 수준이 과도할 경우 H 2 O 2 는 생산 장소에서 멀리 떨어진 세포로 확산되어 산화적 손상과 세포 사멸을 유발할 수 있습니다.
따라서 신호 분자 또는 독성 물질로서의 ROS의 운명은 ROS의 유형, 국소 농도 및 항산화제의 풍부함에 따라 달라집니다.
ROS 조절
ROS 항상성은 정상적인 세포 생존과 적절한 세포 신호 전달에 필요합니다.
낮은 수준의 ROS는 조절된 방식으로 대사 적응, 분화 및 세포 증식을 조절하기 위해 신호 경로를 활성화할 수 있습니다.
정상 세포와 비교하여 암세포는 세포 변형 및 종양 형성에 필요한 세포 신호 전달 경로를 과활성화하는 공간적으로 국소화된 ROS 생성이 증가합니다.그림 2)
ROS 생성이 증가하거나 ROS 제거가 감소하기 때문에 ROS 축적이 발생할 수 있습니다
종양유전자 활성화, 향상된 신진대사 및 저산소증이 ROS 생성을 증가시킬 수 있지만 종양 억제기 손실은 ROS 제거를 감소시킬 수 있습니다
그러나 ROS의 축적은 암세포 사멸을 방지하기 위해 엄격하게 조절되어야 합니다.

그림 2 암세포의 활성산소(ROS) 균형.
암세포는 공간적으로 국한된 ROS 생산 속도를 증가시켜 종양 형성에 필요한 세포 신호 전달 경로를 과활성화합니다.
ROS 축적은 종양유전자 활성화, 종양 억제인자 손실, 대사 활동 증가, 영양소 결핍(예: 낮은 포도당) 또는 제한된 산소(즉, 저산소증)의 결과로 발생할 수 있습니다.
과도한 ROS 수치는 산화적 손상과 세포 사멸을 유발할 수 있기 때문에 암세포는 항산화 능력을 증가시켜 ROS 축적을 조절합니다.
암세포는 전사 인자 NRF2(핵인자 적혈구계 2 관련 인자 2)를 활성화하여 SOD(초산화물 디스뮤타제), PRX(퍼옥시레독신), 글루타티온 퍼옥시다제(GPX) 및 카탈라제(CAT)와 같은 항산화제와 NADPH 생산에 관여하는 유전자; 글루타티온(GSH) 합성 및 이용 유전자; 및 해독 효소의 발현을 증가시킵니다.
ROS 생성과 ROS 제거 사이의 이러한 섬세한 균형은 protumorigenic 신호 전달을 위한 최적의 ROS 수준을 유지합니다.
ROS 항상성을 유지하기 위해 세포는 광범위한 항산화 방어 시스템을 통해 공간적 및 시간적 방식으로 세포 내 ROS 수준을 조절합니다.
철-황 클러스터를 포함하는 단백질을 손상시키고 비활성화시키는 O2-의 능력이 있고 SOD는 O2-를 H2O2로 빠르게 전환시키기 위해 다양한 세포 구획에 위치합니다.
세포 독성을 방지하고 세포 신호 전달을 위한 최적의 범위에서 H2O2 수준을 유지하기 위해, 퍼옥시레독신(PRX), 글루타티온 퍼옥시다제(GPX) 및 카탈라제(CAT)를 포함하여 세포 내 H2O2를 물(H2O)로 전환시키는 수많은 항산화제가 존재합니다. 포유동물 세포에서 PRX의 6가지 동형은 세포질, 미토콘드리아, 소포체 및 과산화소체 전체에 분포되어 있습니다.
다양한 세포 구획에서의 위치와 높은 풍부도는 PRX를 이상적인 H 2 O 2 청소부로 만듭니다.
H 2 O 2 를 해독하기 위해 PRX는 산화제 수용체 역할을 합니다. 그들의 활성 부위 시스테인 잔기는 H 2 O 2 매개 산화를 겪을 수 있습니다.
산화된 PRX는 결과적으로 산화된 TRX가 되는 티오레독신(TRX)에 의해 환원됩니다.
Thioredoxin reductase(TR)와 환원 등가 NADPH는 산화된 TRX를 환원된 형태로 복원합니다.
중요하게는, 진핵생물의 PRX는 두 번째 H 2 O 2 분자가 이를 가역적 으로 sulfinic acid(SO 2 - )로 과산화할 때 일시적으로 비활성화될 수 있으며 sulfiredoxin의 효소 활성에 의해 다시 활성화될 수 있습니다.
GPX 계열의 단백질(GPX1-8)은 또한 세포질과 미토콘드리아에서 H 2 O 2 를 H 2 O 로 전환할 수 있습니다 .
H 2 O 2 를 H 2 O로 환원 하기 위해 GPX는 환원된 글루타티온(GSH)을 이황화 글루타티온(GSSG)으로 산화시킵니다.
산화된 GSSG는 NADPH를 전자 공여체로 사용하는 글루타티온 환원효소에 의해 다시 GSH로 환원됩니다.
CAT는 대부분의 세포의 과산화소체에 보조인자가 없을 때 H 2 O 2 를 H 2 O 및 O 2 로 매우 효율적으로 전환할 수 있습니다 .
따라서 ROS의 세포 내 수준을 조절하기 위해 세포는 협력적이고 강력한 항산화 방어 시스템을 발전시켜 SOD가 O2-를 변화시켜 H2O2를 생성하고 PRX, GPX 및 CAT가 H2O2를 제거하고 H2O를 생성합니다.
언급한 바와 같이 NADPH는 ROS 생성(NOX)과 ROS 해독(감소된 GSH 및 TRX 풀을 보충함으로써)을 모두 촉진할 수 있습니다.
NADPH는 세포질과 미토콘드리아 모두에서 여러 출처에 의해 생성될 수 있습니다.
해당 중간체인 글루코스-6-인산은 오탄당 인산 경로(PPP)를 통해 세포질 NADPH를 생성할 수 있는 반면, 트리카르복실산(TCA) 주기 중간체인 isocitrate and malate는 다른 위치에 있는 isocitrate dehydrogenases (IDH1 and IDH2) and malic enzymes (ME1, ME2, and ME3)의 활성을 통해 각각 세포질 및 미토콘드리아 NADPH를 생성할 수 있습니다.
아미노산 세린과 글리신의 탄소 단위가 엽산 회로로 공급되는 one-carbon 대사는 세포질과 미토콘드리아에서 NADPH를 생성합니다
또한 뉴클레오타이드 합성에는 one-carbon 대사가 필요합니다
따라서 one-carbon 대사가 상향 조절되고 암세포 증식에 필요하다는 것은 놀라운 일이 아닙니다.
특히, 미토콘드리아 one-carbon 대사를 통한 세린 이화작용은 종양 저산소증 동안 산화 환원 균형을 유지하여 암세포 증식을 허용합니다.
Myc로 형질전환된 세포의 저산소증 동안 , 전사 인자인 저산소증 유도 인자(HIF)는 Myc와 협력하여 미토콘드리아 one-carbon 대사 효소인 SHMT2(세린 하이드록시메틸트랜스퍼라제 2)의 발현을 유도합니다.
이는 NADPH 생성을 증가시키고 세포 항산화 능력을 유지하여 저산소증으로 인한 미토콘드리아 ROS(mROS) 증가를 상쇄합니다.
SHMT2의 녹다운은 항산화제 N- 아세틸 시스테인(NAC)에 의해 구제되는 효과인 세포 사멸을 증가시키는 것으로 밝혀졌기 때문에 과도한 수준의 ROS는 세포 독성 및 사멸을 유도할 가능성이 높습니다
세포는 항산화 단백질의 발현을 증가시키고 산화 환원 균형을 유지하기 위해 (NRF2) 전사 인자를 활성화합니다.
산화 스트레스 동안 NRF2는 안정화(분해되지 않음)되고 핵으로 이동하여 GPX , PRX 및 CAT, GSH 합성 및 이용에 관여하는 효소; 해독 효소와 같은 항산화제를 비롯한 수많은 유전자의 발현을 유도할 수 있습니다 .
또한 NRF2 활성화는 PPP 및 세린 생합성 경로에서 효소를 조절하여 NADPH 생성을 촉진할 수 있습니다.
Kras 및 Myc 와 같은 종양 유전자의 돌연변이뿐만 아니라 KEAP1 및 NRF2의 돌연변이는 NRF2 를 활성화하는 것으로 나타났습니다
암 세포에서 NRF2 매개 유전자 발현으로 인한 향상된 ROS 소거는 사망 유도 JNK/p38 경로의 ROS 매개 활성화와 돌이킬 수 없는 산화 손상을 방지함으로써 ROS 항상성을 유지할 가능성이 있습니다.
실제로 NRF2는 암세포 증식과 종양 형성에 필수적인 것으로 나타났습니다
따라서 NRF2의 손실은 산화 스트레스로 인한 암세포 사멸을 촉진하여 종양 부담을 줄입니다.
또한, 종양 억제제는 정상 세포에서 항산화 기능을 할 수 있습니다. 종양 억제인자 BRCA1은 결합 파트너인 NRF2의 안정성과 활성화를 촉진하여 ROS 축적을 억제합니다.
미토콘드리아 종양 억제인자 시르투인 3(SIRT3)는 SOD2를 탈아세틸화하여 소거 기능을 조절합니다.
SIRT3가 없으면 SOD2 활성이 감소하여 ROS 축적, HIF-1α 단백질 안정화 및 종양 발생의 결과를 나타냅니다.
종양 억제인자 p53은 SOD2, GPX1 및 CAT를 포함한 다양한 항산화 유전자의 발현을 조절함으로써 ROS 축적을 억제하는 것으로 나타났습니다
그러나 인간 암의 50% 이상에서 관찰되는 바와 같이 p53이 손실되거나 돌연변이되면 ROS 축적이 발생할 수 있어 원발성 신호 전달 및 세포 변형이 가능합니다.
항산화제 NAC의 식이 보충은 p53 결핍 마우스 모델에서 종양 성장을 감소시켰습니다
따라서 p53은 부분적으로 ROS의 조절을 통해 종양 억제 기능을 매개할 수 있습니다.
또한, 대사 유전자 TIGAR 의 유도를 통해 p53은 항산화 기능을 조절할 수 있습니다
해당 작용의 음성 조절자인 TIGAR는 과당-2,6-이인산 fructose-2,6-bisphosphate 농도를 낮추고 포도당 탄소를 PPP로 리디렉션하고 세포질 NADPH 생성을 증가시켜 ROS 축적을 억제합니다.
흥미롭게도, 세포 사멸의 비세포사멸 형태인 p53 매개 ferroptosis는 시스테인-글루타메이트 항포터 SLC7A11의 p53 의존적 전사 억제를 포함하여 시스테인 흡수 감소 및 후속 GSH 합성 및 상승된 ROS 수준을 초래합니다.
NADPH의 풍부함은 또한 ferroptosis를 유발하는 화합물에 대한 감수성과 반대의 상관관계가 있는 것으로 밝혀졌습니다.
따라서 유방암과 간암을 포함한 여러 인간 암이 SLC7A11을 과발현하여 항산화 능력을 유지하고 ferroptosis를 피하는 것은 놀라운 일이 아닙니다
신호 분자로서의 ROS
H 2 O 2 - 매개 시스테인 산화
H 2 O 2 는 PTEN, PTP1B, PP2A를 포함한 포스파타제의 중요한 시스테인 티올 그룹을 가역적으로 산화시켜 그들의 활성을 억제하고 PI3K/Akt 생존 경로의 활성화를 촉진하는 것으로 나타났습니다
더욱이, 프롤릴 하이드록실라제 도메인 단백질 2(PHD2)의 H 2 O 2 매개 산화는 저산소 상태 동안 HIF-1α 단백질의 발암성 안정화를 초래할 수 있습니다.
표적 단백질 의 티올레이트 음이온(S - )은 H 2 O 2 에 의해 가역적으로 산화되어 설펜계 형태(SO - )가 될 수 있으며, 이는 후속적으로 단백질 형태와 활성을 변경하고 비가역적 시스테인 산화로부터 표적 단백질을 보호합니다.
H 2 O 2 - 의존 신호 변환
광범위하고 다중 구획 세포 항산화 시스템을 고려할 때 표적 단백질이 H 2 O 2 에 의해 구체적이고 효율적으로 어떻게 산화되는지는 불분명합니다 .
H 2 O 2 에 의한 표적 단백질의 직접적인 산화는 반응성이 높은 항산화제 존재에서는 거의 일어나지 않는 것 같습니다.
H 2 O 2 에 의한 표적 단백질 산화를 촉진하는 한 메커니즘 은 H 2 O 2 신호 를 감지하고 변환하는 ROS 소거 효소의 능력에 의존할 수 있습니다.
산화환원 릴레이라고 하는 이 메커니즘은 소거 효소(예: PRX 또는 GPX)가 H 2 O 2 산화를 받고 산화를 표적 단백질로 전달하는 2 단계 과정입니다 .
인산화와 같은 번역 후 변형은 소거 효소를 비활성화하는 또 다른 방법입니다.
성장 인자 수용체 자극은 NOX 유래 H 2 O 2 생성과 Src 키나아제 활성화를 증가시켜 PRX1의 인산화 및 비활성화를 유발합니다.
H 2 O 2 는 Src를 추가로 활성화하고 포스파타제를 억제하여 PRX1의 공간적 및 시간적 비활성화를 유지하여 국소화된 H 2 O 2 의존적 신호 전달을 가능하게 합니다.
아마도 유사한 메커니즘이 미토콘드리아에서 발생하여 mROS가 Src 계열 구성원인 막간 공간에 국한된 Lyn과 Syk 키나아제 경로를 활성화하여 전사, 대사 및 세포 주기 조절과 같은 세포 과정을 제어합니다.
또한, 티올 함유 화합물 GSH 또는 환원된 TRX가 산화된 잔유물을 환원된 상태로 다시 재활용하고 표적 단백질에 신호를 전달하는 것을 생각할 수 있습니다.
TRX1은 AMPK의 이황화 환원을 촉매하여 허혈 동안 AMPK 산화 응집 및 비활성화를 방지하는 것으로 나타났습니다
또한, 소거 효소의 H 2 O 2 산화는 표적 단백질의 해리 및 활성화를 초래할 수 있다.
ROS는 PROTUMORIGENIC SIGNALING을 촉진합니다.
1990년대 중반의 선구적인 연구는 ROS가 종양 형성을 촉진한다는 것을 보여주었습니다.
발암성 Ras 및 성장 인자 신호 전달은 PI3K/Akt/mTOR 및 MAPK/ERK 신호 전달 캐스케이드의 H 2 O 2 활성화를 유도할 수 있습니다 (그림 3)
Ras의 발암성 돌연변이는 증식을 향상시키기 위해 NOX4에서 ROS 생성을 증가시킬 수 있습니다.
ROS는 PI3K/Akt 신호전달의 음성 조절자인 포스파타제 PTEN 및 PTP1B를 산화 및 비활성화함으로써 PI3K/Akt/mTOR 생존 경로를 과활성화할 수 있습니다.
PTEN은 교모세포종, 흑색종, 전립선암, 유방암 및 자궁내막암을 포함한 수많은 인간 암에서 자주 비활성화됩니다.
따라서 ROS 유도 PI3K/Akt 생존 경로는 대부분의 암에서 활성화되는 것으로 나타났습니다
또한 Akt의 발암성 활성화는 ROS 생성을 증가시켜 암세포의 생존과 증식을 더욱 촉진할 수 있습니다.
ROS는 또한 MAPK 포스파타제를 산화 및 비활성화하여 성장 인자 수용체 활성화 및 MAPK/ERK 증식 촉진 신호를 유도합니다.
미토콘드리아 ROS 는 MAPK/ERK 경로를 통해 Kras가 유도하는 고정과 무관한 폐암 세포의 성장을 조절합니다.
또한, ROS를 감소시키는 미토콘드리아 호흡 사슬의 완전한 파괴는 종양 형성을 감소시키는 것으로 나타났습니다.연구에서 mROS가 종양형성에 더 관련이 있음을 나타냅니다
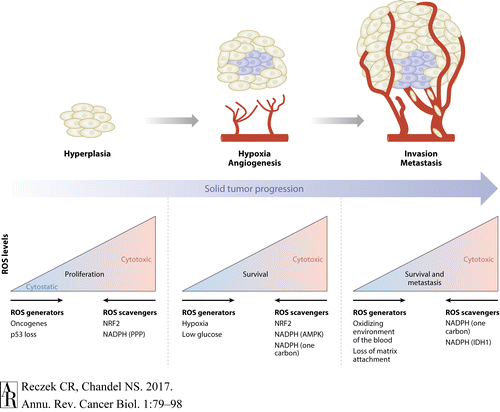
그림 3 고형 종양 진행 및 활성산소종(ROS).
종양유전자 활성화 및 종양 억제인자 손실은 ROS를 생성하여 증식 촉진 신호 및 증식을 촉진할 수 있습니다.
ROS의 축적을 방지하기 위해 암세포는 항산화제 마스터 레귤레이터 NRF2(핵인자 적혈구계 2 관련 인자 2)를 상향 조절하고 오탄당 인산 경로(PPP)를 통한 플럭스를 증가시켜 NADPH를 생성합니다.
고형 종양이 성장함에 따라 혈관계 접근성이 제한됩니다. 따라서 종양 내의 영역은 저산소 상태(즉, 낮은 산소)가 되고 포도당이 결핍되어 ROS 수준이 추가로 증가합니다.
포도당 수치가 낮을 때 PPP는 더 이상 NADPH 생산을 증가시키는 주요 경로가 아닙니다.
대신 암세포는 AMPK와 1탄소 대사 경로를 활성화하여 NADPH 생성을 강화하고 산화환원 항상성을 유지합니다.
ROS 수치는 암세포가 세포외기질에서 분리되어 기저막을 침범하여 혈액의 산화 환경으로 들어갈 때 더욱 증가합니다.
고정 독립 성장과 원격 전이를 지원하기 위해 암세포는 IDH1(isocitrate dehydrogenase 1) 의존적 환원성 카르복실화 및 1탄소 대사 경로를 통해 NADPH 생성을 증가시켜 항산화 능력을 높입니다.
암세포의 산화환원 균형을 교란시킴으로써 종양의 진행과 전이를 예방할 수 있습니다.
ROS는 또한 NF-κB 및 NRF2의 활성화를 통해 종양 세포 생존을 촉진합니다. 이는 ROS 매개 암세포 사멸을 피하기 위해 항산화제의 발현을 상향 조절하는 전사 인자입니다.
최근 연구에서 발암성 Kras 유래 mROS는 PKD1을 통해 NF-κB를 활성화하여 증식성 EGFR 신호를 상향 조절하여 췌장 전암 병변을 형성합니다.
더욱이, 종양 억제인자 APC의 손실은 최근 RAC1 및 TIGAR의 상향 조절을 통해 비정상적인 Wnt 신호 전달 및 장 증식 intestinal proliferation 을 매개하는 것으로 나타났습니다.
역설적으로 RAC1은 NOX 활성화를 유도하여 ROS 생성을 증가시키지만 TIGAR는 세포질 NADPH 수준을 증가시켜 ROS를 감소시킵니다. 최적의 장 증식을 촉진하기 위해 TIGAR 유도 NADPH가 항산화 기능을 회복시켜 손상된 ROS 풀이 소거되어 RAC1이 증식 촉진 신호 ROS 풀을 증가시키는 것으로 생각됩니다.
또한, ROS는 종종 불량한 예후와 관련된 상호 관련된 과정인 종양 혈관신생 및 전이를 촉진합니다. 고도로 증식하는 종양이 혈액 공급을 초과할 때, 고형 종양 내의 영역은 저산소 상태가 되고 포도당이 결핍되어 ROS 생성을 촉진합니다. 이 가혹한 종양 미세 환경에서 생존하고 증식하기 위해 암세포는 HIF 안정화, AMPK 및 1탄소 대사 경로의 활성화를 포함한 대사 변화를 거쳐 NADPH 생성을 강화하고 산화 환원 균형을 유지합니다
저산소증으로 유도된 mROS는 PHD2 활성을 억제하여 산소에 민감한 HIF-α 소단위를 안정화시킵니다.
정상산소 조건(충분한 산소)에서 HIF-α는 PHD2 매개 히드록실화 후 분해됩니다.
그러나 저산소 상태에서 HIF-α 단백질은 HIF-β 소단위와 이합체화되고 핵으로 전위되어 혈관 내피 성장 인자 및 세포 생존, 해당 작용 및 전이에 관여하는 유전자와 같은 혈관 신생 유전자의 발현을 유도합니다.
ROS 의존 메커니즘을 통한 HIF-α 안정화는 특정 암세포의 종양 형성을 촉진하는 것으로 나타났습니다.
낮은 포도당 공급으로 인해 PPP 의존적 NADPH 생산이 감소합니다. 따라서 고형 종양에서 ROS 수준이 증가합니다. 항산화 능력을 높이고 ROS 매개 종양 세포 사멸을 방지하기 위해 AMPK가 활성화되어 NADPH 생성을 촉진하고 NADPH 소비가 필요한 동화 과정을 방지합니다.
실제로, AMPK의 손실은 발암성 형질전환을 예방하는 것으로 나타났습니다
또한 저산소 상태에서 단일 탄소 대사 효소 SHMT2의 HIF 의존적 상향 조절은 미토콘드리아 세린 이화 작용과 NADPH 생성을 촉진합니다.
전이는 세포-세포 접착의 상실을 필요로 하며, 그 결과 암세포가 원발성 종양에서 해리되고 기저막으로 돌파됩니다.
ROS는 수많은 신호 전달 경로(예: MAPK 및 PI3K/Akt 경로)와 전사 활성(예: HIF 및 달팽이)을 조절하여 암세포의 이동과 침입을 향상시키는 것으로 나타났습니다.
NOX 유래 ROS는 종양 세포가 침입하기 위해 사용하는 원형질막 돌출부인 invadopodia의 형성을 촉진합니다.
또한, ROS 의존성 v-Src 산화 및 후속 활성화는 Src-형질전환된 세포의 침입 가능성 및 고정 및 성장 인자-독립적 성장을 향상시키는 것으로 나타났습니다.
Anoikis는 기질 박리로 인한 세포자멸사로서 전이를 예방하는 데 중요한 역할을 합니다. ROS는 Src의 산화 및 활성화를 통해 암세포에 아노이키스 저항성을 부여하여 구성적이고 리간드 독립적인 EGFR 활성화 및 생존 신호전달을 유도하는 것으로 나타났습니다.
복합체 I 활성을 손상시키는 미토콘드리아 DNA의 돌연변이로 인한 상승된 ROS 수준은 또한 마우스 및 인간 종양 세포주의 전이를 촉진하는 것으로 나타났습니다.
이러한 발견은 세포 사멸에 대한 역치 미만의 ROS 수준의 증가가 국소 침습 및 전이를 촉진하기 위해 종양 세포의 구조적 변화 및 적응 반응을 조절하는 데 필요함을 시사합니다.
ROS는 항종양유전자 신호를 촉진합니다.
ROS 수치가 너무 높으면 세포 주기 정지, 노화 및 암세포 사멸이 발생할 수 있습니다
ROS는 ASK1/JNK 및 ASK1/p38 신호 전달 경로의 활성화를 통해 세포 사멸을 촉진합니다.
비활성 상태에서 ASK1은 감소된 TRX와 상호 작용합니다. 그러나 TRX의 H 2 O 2 매개 산화는 ASK1 해리 및 활성화를 유발하여 다운스트림 (MKK)4/MKK7/JNK 및 MKK3/MKK6/p38 경로의 지속적인 활성화를 통해 항-세포자멸 인자의 억제를 촉발합니다.
p38 및 JNK 경로 구성요소의 비활성화 돌연변이는 수많은 인간 종양에서 관찰되었으며, 이는 이러한 신호 전달 경로가 암세포 사멸을 촉진한다는 것을 시사합니다.
또한, p38 MAPK는 종양 억제 인자로 특징지어지며 발암성 Hras 악성 형질전환을 억제합니다
JNK 및 p38 신호 전달 경로의 ROS 매개 활성화는 또한 세포 주기 정지를 유도하여 암세포의 성장과 분열을 방지할 수 있습니다.
종양 세포가 세포외 기질에서 분리되어 기저막을 침범함에 따라 ROS 수준이 증가하고 감소된 GSH 풀이 감소합니다.
혈액의 산화 환경은 순환하는 많은 암세포의 생존과 증식을 방지합니다.
따라서 과도한 ROS 수준은 원격 전이를 억제합니다
고정 독립 성장을 지원하고 전이 중 세포 사멸을 피하기 위해 암세포는 항산화 능력을 증가시키기 위해 대사 변화를 겪습니다.
특히, 세포질 IDH1 의존적 환원성 카르복실화는 미토콘드리아 NADPH의 풍부함을 증가시키고 mROS 수준을 낮추어 고정 독립 성장을 촉진합니다.
또한, 인간 흑색종 세포는 원격 전이를 촉진하기 위해 엽산 매개 1탄소 대사 경로의 NADPH 생성 효소를 필요로 합니다.
따라서 이러한 ROS 완화 경로를 교란시키는 것은 암세포 증식 및 전이를 억제하기 위한 실행 가능한 치료적 접근이 될 수 있습니다.
암 치료를 위한 표적화 ROS
암에서 ROS의 두 가지 얼굴, 즉 pro- 및 antitumorigenic이 있습니다.
ROS의 증가는 암 세포 증식과 생존을 촉진하는 국소 신호 전달을 허용하지만, ROS 소거의 증가는 원격 ROS 매개 산화 손상을 감소시키고 ROS 항상성을 유지합니다.
암세포 증식을 방지하기 위한 ROS 수준 감소
그들의 protumorigenic 신호 전달 기능을 통해 ROS는 종양 형성에 인과 관계가 있습니다. 따라서 세포 내 ROS 수준을 낮추고 암세포 증식을 예방하는 방법을 개발하는 것이 매력적입니다. 잠재적인 암 치료법 중 하나는 항산화제를 사용하여 ROS 수준을 낮추는 것입니다.
Myc 의존성 림프종 모델 생쥐에서, 항산화 제 NAC 및 비타민 C는 HIF-1α 단백질 발현을 감소시킴으로써 종양 억제하는 것으로 나타났다
또한 SOD3의 과발현은 생체 내 유방암 전이를 감소시켰습니다
이러한 결과는 유망하지만 여러 대규모 전향적 임상 연구에서 β-카로틴, 비타민 A 또는 비타민 E를 포함한 식이 항산화제 보충제가 암 발병률을 증가시키는 것으로 나타났습니다
이러한 발견과 일치하게, NAC와 비타민 E 보충제는 Kras 및 Braf로 유도된 폐암 마우스 모델에서 종양 발달과 사망률을 현저하게 가속화 했습니다.
NAC 투여는 또한 발암성 Braf/Pten -null 마우스 에서 흑색종 전이를 증가 시켰 습니다.
이러한 혼합된 결과는 암 치료에 항산화제를 사용하는 것의 복잡성을 보여줍니다.
식이 항산화제가 protumorigenic 신호 ROS 풀을 효율적으로 청소하지 않고 대신 생산 위치에서 멀리 떨어진 ROS의 독성을 감소시켜 종양 진행을 촉진할 수 있습니다.그림 4).
이를 극복하기 위해 protumorigenic 신호와 관련된 국소적으로 생성된 ROS에 접근하고 해독할 수 있는 표적 항산화제(예: 미토콘드리아 표적 항산화제)를 사용하는 것이 유익할 수 있습니다.
미토콘드리아 표적 O 2 - 스캐빈저 MitoTEMPO는 인간 및 쥐 모델에서 종양 세포 이동을 차단하고 자발적인 종양 전이를 억제하는 것으로 나타났습니다
또한, 미토콘드리아를 표적으로 하는 항산화제 MitoQ는 Kras에 의해 유도된 췌장 전암 병변 의 형성을 감소시켰으며 , 이는 mROS가 원발성 신호를 전파함을 시사합니다.
흥미롭게도, 미토콘드리아 H 2 O 2 생성 에 대한 635,000개의 소분자에 대한 최근의 고처리량 스크린은 복합체 III O 2 - 생성 을 특이적으로 억제 하고 미토콘드리아의 생체 에너지 기능을 손상시키지 않으면서 HIF-1α 단백질 안정화를 방지 하는 3가지 화합물을 밝혀냈습니다
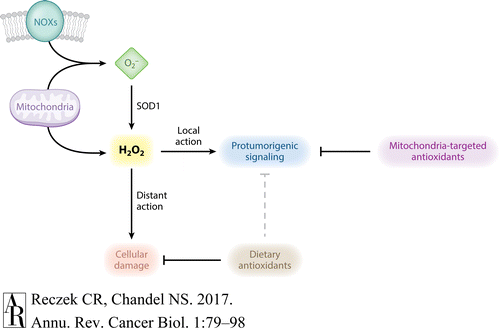
그림 4 활성산소종(ROS)의 항산화 요법과 국소화.
미토콘드리아와 막 결합 NADPH 산화효소(NOX)는 슈퍼옥사이드(O 2 - )를 생성 하며, 이는 세포질 슈퍼옥사이드 디스뮤타제 1(SOD1)에 의해 과산화수소(H 2 O 2 ) 로 전환될 수 있습니다 .
미토콘드리아 유래 H 2 O 2 는 미토콘드리아 밖으로 유출되어 세포질 H 2 O 2 풀에 기여할 수 있습니다 .
생산 부위 근처의 H 2 O 2 는 국소적으로 작용할 수 있고 주변의 표적 단백질을 산화시켜 protumorigenic 신호 전달을 촉진할 수 있습니다.
그러나 H 2 O 2 의 수준이 높으면 H 2 O 2 는 세포의 먼 부분으로 이동하여 단백질, 지질 및 DNA에 산화적 손상을 일으킬 수 있습니다.
종양 형성에서 ROS의 인과적 역할을 감안할 때, ROS 수준을 낮추기 위한 항산화 요법은 매력적입니다.
식이 항산화제 보충제는 암 발병률을 줄이는 데 실패했습니다.
왜냐하면 그것이 손상을 유발하는 국소적인 원발성 신호인 H 2 O 2 가 아니라 멀리 떨어져 있는 H 2 O 2 를 억제하기 때문 입니다.
그러나 H 2 O 2 생산 의 근원에 접근하는 미토콘드리아를 표적으로 하는 항산화제 는 protumorigenic 신호와 관련된 H 2 O 2를 낮출 수 있습니다.
그리고 암세포의 증식을 억제합니다. 따라서 표적 항산화 요법은 암 치료에 도움이 될 수 있습니다.
산화 스트레스로 인한 암세포 사멸 촉진을 위한 ROS 수치 증가
암세포는 정상 세포에 비해 ROS 수준이 높기 때문에 ROS 생성 전략에 의한 산화 스트레스로 인한 세포 사멸에 잠재적으로 더 취약합니다. ROS 생산을 증가시키거나 ROS 소거를 감소시켜 ROS 수준을 높이면 정상 세포는 보존하면서 암 세포를 한계 너머로 밀어낼 수 있습니다. 최근 고용량의 항산화제 비타민 C는 유전자형 선택적 항암제인 pro-oxidant(ROS 생성기)로 작용하여 Kras 및 Braf 돌연변이 결장암 세포 의 세포 사멸을 유도하는 것으로 나타 났습니다
게다가, 소분자를 이용한 연구에서 ROS의 정상 상태 수준을 증가시키는 Kras 돌연변이 암 세포 의 선택적 킬러인 란페리손(lanperisone) 화합물을 확인했습니다.
GSH와 빠르게 결합하고 GSH 풀을 고갈시키는 화합물은 산화 스트레스로 인한 암세포 사멸을 촉진합니다.
또한, GSH 합성에 필요한 효소인 글루타메이트 시스테인 리가아제(GCL)의 억제제인 부티오닌 설폭시민(BSO)은 GSH를 고갈시키고 항암 활성을 나타냅니다.
또한 최근 연구에서 포도당 유사체 2-deoxy- d -glucose (2DG) 와 함께 전달된 BSO 는 시험관 내 및 생체 내에서 대사적으로 재프로그래밍된 돌연변이 Kras 복사 이득 특이적 폐암 세포 의 세포 사멸을 선택적으로 유도했습니다.
더욱이, 편향되지 않은 스크린에서 확인된 소분자 piperlongumine은 감소된 GSH의 풍부함을 감소시켜 ROS 수준과 암세포의 선택적 세포 사멸을 향상시켰습니다.
이 치료적 접근은 종종 화학요법 및 이온화 방사선에 내성이 있는 암 세포의 고도로 종양 발생성인 하위 집단인 암 줄기 세포(CSC)를 표적으로 하는 데 특히 유익할 수 있습니다.
CSC는 부분적으로 증가된 PPP 의존적 NADPH 생산과 GSH와 같은 항산화제의 과발현으로 인해 일반 암세포 및 비종양형성 세포에 비해 낮은 수준의 ROS를 가지고 있습니다.
예를 들어, CD44v를 발현하는 인간 위장 CSC는 시스테인-글루타메이트 교환 수송체를 안정화시키고 GSH의 합성을 증가시키는 것으로 나타났습니다
CSC의 강화된 항산화 능력은 CSC 자가 재생 및 분화를 허용할 뿐만 아니라 CSC가 방사선 및 기존 화학 요법에 의해 유도된 ROS 매개 산화 손상 및 세포 사멸을 피할 수 있도록 합니다.
실제로, BSO로 GSH 합성을 억제하면 유방 CSC의 집락 형성 능력과 방사선 저항성이 감소했습니다.
또한, CSC는 PI3K/Akt/mTOR 및 Notch 경로와 같은 수많은 ROS 의존적 신호 전달 경로를 상향 조절함으로써 생존 및 줄기와 유사한 특성을 유지합니다.
낮은 ROS 수준이 CSC의 생존을 지원하는 것으로 보이기 때문에 ROS 수준을 증가시키는 항암 요법은 CSC의 사망을 유발하는 데 효과적일 수 있습니다.
GSH 풀을 낮추는 것 외에도 TRX, TR 및 SOD1의 억제제를 포함한 다른 항산화제가 암세포를 선택적으로 죽이는 표적으로 등장했습니다.
또한, ROS 생성 CDK4 및 CDK6 억제제와 함께 항산화제 CAT를 표적으로 하는 것은 ROS 수준을 역치 이상으로 높이고 췌관 선암종 세포 성장을 감소시키는 것으로 나타났습니다.
또한 항산화제 마스터 레귤레이터인 NRF2를 억제하는 것이 효과적인 항암 요법임을 입증할 수 있습니다. 그러나 NRF2 억제제 개발의 과제는 특이성입니다
따라서 특정 NRF2 조절 NADPH 생성 대사 경로를 표적으로 하는 것이 항암 치료에 더 효과적인 접근 방식일 수 있습니다
수많은 종양 유형에 대한 RNA 프로파일링은 미토콘드리아 1탄소 대사 효소인 methylenetetra-hydrofolate dehydrogenase 2가 정상적인 성인 증식 세포에 비해 암세포에서 높게 발현된다는 것을 보여주었습니다.
따라서 미토콘드리아 1탄소 대사 경로를 표적으로 하는 것은 특히 치료 옵션이 제한된 저산소 종양에서 선택적이고 효과적인 항암 요법을 생산할 수 있습니다.
암세포는 증식을 촉진하기 위해 빠르게 세린을 소비하기 때문에 영양소 가용성을 조절하는 것은 유망한 치료 전략이 될 수 있습니다.
세린 기아 동안 정상세포의 p53의 존재는 p21 의존적 세포 주기 정지 및 새로운 세린이 세포 항산화 능력을 유지하고 산화 스트레스를 방지하기 위해 GSH 합성으로 리디렉션되도록 허용했습니다.
이 발견은 세린 고갈이 많은 암 유형에서 관찰되는 일반적인 유전적 변화인 p53 결핍 종양을 치료하는 데 사용될 수 있음을 시사합니다.
또한 ROS 생성 물질과 ROS 소거 억제제를 결합하여 암세포 적응과 화학 요법에 대한 내성을 줄이는 것이 도움이 될 수 있습니다.
결론
게놈 불안정과 종양 형성을 촉진하는 독성 화학 종에서 pro- 및 antitumorigenic 신호 이벤트에 참여할 수 있는 중요한 신호 분자에 이르기까지 암의 ROS에 대한 이해가 바뀌었습니다. 종양 형성을 촉진하는 ROS의 인과적 역할과 항산화제가 종양 억제인자라는 널리 알려진 믿음을 감안할 때, ROS 조작 전략은 이전에 항산화 요법에 초점을 맞추었습니다. 그러나 대부분의 임상 시험에서 식이 항산화제의 실패로 대체 항암 치료 접근법이 등장했습니다
protumorigenic 신호는 ROS 생성 부위 근처에서 발생하기 때문에 ROS 생성 소스를 표적으로 하는 항산화제 또는 억제제를 사용하면 종양 형성을 예방할 수 있습니다.
게다가, 초기에는 반직관적이지만, 활성산소 생성을 증가시키거나, ROS 소거를 감소시키거나, 또는 둘 다 하는 산화촉진제 암 요법은 정상 세포에 비해 암세포에서 더 높은 수준의 ROS 생성을 고려할 때 매력적인 접근 방식입니다.
산화촉진 요법의 결과로 ROS 축적은 암세포가 ROS 항상성을 잃어 산화 스트레스로 인한 세포 사멸로 균형을 전환하기에 충분히 높을 수 있습니다.
이 접근법을 사용할 때 정상 세포, 특히 줄기 세포가 ROS 수준에 민감하다는 점을 고려하는 것이 중요합니다
따라서 산화 환원 균형을 유지하기 위해 암세포가 선택적으로 사용하는 특정 항산화 경로를 식별하고 표적화하는 것이 중요합니다. 암에서 ROS 신호 전달의 분자 메커니즘에 대한 더 나은 이해와 특정 ROS 표적의 식별은 암 치료를 위한 새로운 치료 방법을 제공할 수 있습니다.
'정보' 카테고리의 다른 글
| ⚡신경과 질환(대한약사회지) (0) | 2022.01.12 |
|---|---|
| ⚡영양제 구입 (0) | 2021.11.12 |
| 암 예방 및 치료에서 ROS의 양날 역할 (0) | 2021.11.07 |
| 암 치료와 ROS (0) | 2021.10.18 |
| 암 화학 요법에서 식이 항산화제의 수수께끼 (0) | 2021.10.18 |