2019
Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells
https://www.mdpi.com/2072-6694/11/7/916/htm
Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells
The transformation of normal cells to the cancerous stage involves multiple genetic changes or mutations leading to hyperproliferation, resistance to apoptosis, and evasion of the host immune system. However, to accomplish hyperproliferation, cancer cells
www.mdpi.com
정상 세포가 암 단계로 전환되는 것은 다중 유전적 변화 또는 돌연변이를 수반하여 과다증식, 세포자멸사에 대한 저항성 및 숙주 면역계의 회피를 초래합니다.
그러나 과증식을 달성하기 위해 암세포는 산화적 해당과정과 세포질의 산성화를 포함하는 심오한 대사 재프로그래밍을 거쳐 미토콘드리아 막의 과분극을 초래합니다.
과거 약물 개발 연구의 대부분은 암세포에서 세포 사멸을 유도하기 위해 DNA 복제, 복구 및 튜불린 중합을 표적으로 하는 데 중점을 두었습니다.
불행히도, 이들은 암 선택적인 표적이 아닙니다.
최근 연구자들은 암세포 사멸을 유도하기 위한 선택적 표적으로 활용될 수 있는
암세포의 대사,
미토콘드리아 및
산화 스트레스 취약성에 초점을 맞추기 시작했습니다.
암세포에서 미토콘드리아 막의 과분극은 세포 사멸 효과를 유도할 수 있는 미토칸에 의해 이루어질 수 있습니다. 여기에서는 암세포에 대해 선택적 독성을 나타내는 최근의 미토콘드리아 선택적 항암 화합물(미토칸)에 대해 논의합니다.
증가된 산화 스트레스는 또한 암세포에서 선택적으로 세포 사멸을 유도하는 데 매우 효과적인 것으로 나타났습니다.
이 산화 스트레스는 미토콘드리아 기능 장애로 이어질 수 있으며, 이는 차례로 더 많은 활성 산소 종(ROS)을 생성합니다.
이것은 미토콘드리아 기능 장애와 ROS 생성의 악순환을 만들어 비가역적으로 세포 자살로 이어집니다
1. 미토콘드리아와 암의 배경
암세포는 많은 유전적 돌연변이의 축적을 통해 최적이 아닌 조건에서 증식하는 능력이 향상되어 악명이 높습니다. 그들은 어려운 조건에서 적응하고 작동하기 위해 대사 및 면역원성 경로를 조작할 수 있습니다
실제로, 암세포의 대사 재프로그래밍은 암의 특징이자 이 질병과 싸우기 위해 표적이 될 수 있는 잠재적인 취약성으로 확인되었습니다
세포 사멸의 복잡하고 생리학적인 과정인 아폽토시스는 원치 않는 세포와 손상된 DNA를 가진 세포를 모두 제거합니다.
그렇게 함으로써, 이 과정은 비정상 세포의 발달에 대한 보호 역할을 할 수 있습니다.
세포 사멸은 DNA 손상, 산화 스트레스, 성장 인자 결핍 및 미토콘드리아 탈분극에 의해 유발될 수 있습니다
내부 경로에서 DNA 손상과 같은 내부 신호는 일련의 사건으로 이어질 수 있으며, 그 결과 Bcl-2 계열의 단백질이 활성화되고, 이는 차례로 Bax 유사 단백질을 활성화하여 막을 투과하고 시토크롬 c를 방출합니다
시토크롬 c 방출은 카스파제 단백질의 활성화를 유도하여 세포자멸사를 유도합니다.
외부 경로에서 종양 괴사 인자(TNF)와 같은 외부 신호는 카스파제 활성화 및 세포 사멸로 이어질 수 있습니다
불행하게도, 암세포는 세포 사멸에 저항하고 세포 외부로 약물을 내보내 많은 화학 요법에 대한 내성을 발달시킵니다.
이것은 pro-apoptotic protein을 파괴하고, caspase의 기능을 감소시키며, 사멸 수용체 신호 전달을 손상시킴으로써 달성됩니다
DNA 손상제(예: 시스플라틴, 독소루비신 또는 5-플루오로우라실) 및 튜불린 변형제(예: 파클리탁셀)를 포함하는 암 치료제는 암세포에서 세포자멸사를 유도하기 위해 개발되었습니다.
그러나 이들 약물은 비선택적 성질과 건강한 조직에 대한 독성이 매우 커서 한계가 있다. 또한, 암세포는 이러한 약물에 대한 내성을 발달시키고 환자는 암에 큰 영향을 미치지 않으면서 심각한 부작용을 경험할 수 있습니다
또한, 이러한 유전독성 치료는 정상 조직의 DNA 손상을 일으켜 암이 더 발전할 위험을 증가시킬 수 있습니다.
따라서 우리는 본질적으로 암을 치료하려는 시도에서 암 유발 화합물을 사용하고 있습니다.
적대적인 조건에서 증식하기 위해 암세포는 에너지 요구를 충족시키기 위해 대사 재프로그래밍을 겪습니다.
1920년대에 Otto Warburg는 '산화적 해당작용'의 결과로 산소가 있는 상태에서 암세포가 과도한 젖산을 생성하는 것을 설명하는 'Warburg 효과'라고 명명한 것을 관찰했습니다
그러나 이것은 암세포가 미토콘드리아 손상과 대사 비효율을 가지고 있다는 오해를 불러일으켰고, 이로 인해 암세포가 에너지 생산을 위해 과도한 해당과정에 의존하게 되었습니다
이와는 대조적으로, 많은 연구에서 미토콘드리아와 미토콘드리아 호흡이 많은 암에서 손상되지 않은 상태로 남아 있음을 보여주었습니다
흥미롭게도 난소암에서 암세포의 미세 환경은 대사 재프로그래밍에 직접적인 영향을 미칩니다
정상 산소 수준에 노출된 종양 회전 타원체의 말초 세포는 호기성 해당 작용에 크게 의존하고 증식성 세포였습니다. 대조적으로, 내부 세포는 산소와 포도당에 대한 열악한 접근으로 혈관이 잘 형성되지 않아 세포 정지와 ATP 생산의 대부분을 미토콘드리아 산화적 인산화(OxPhos)에 크게 의존하게 되었습니다
이 연구는 표준 화학요법이 증식성 세포를 죽이는 데 효과적이었지만 정지된 세포 집단은 내성이 있고 종양 재생을 유발한다는 사실을 강조합니다.
암을 적절하게 퇴치하기 위해서는 완전한 종양 제거를 보장하기 위해 미토콘드리아 취약성을 표적으로 하는 다면적 접근을 고려해야 합니다.
사실, 최근 관찰에 따르면 미토콘드리아는 대사 재프로그래밍, 산화 신호 전달, 활성 산소 종 생성 및 대사 산물 생성을 통해 실제로 종양 형성을 지원하고 중요한 역할을 합니다
암세포는 건강한 세포에 비해 대사 요구와 항산화 방어가 더 높습니다.
그들은 에너지 요구 사항을 충족하기 위해 호기성 해당 작용에 크게 의존하고 결과적으로 요구 사항을 충족하기 위해 포도당 수송체를 상향 조절합니다
또한, 호기성 해당작용은 다량의 젖산과 피루브산을 생성하여 암세포의 세포질에서 산성도를 증가시킵니다.
따라서 이들의 미토콘드리아는 정상 세포의 미토콘드리아에 비해 과분극되어 있다
이 과분극은 또한 세포 내 Ca 2+ 수준의 증가와 항-세포자멸사 Bcl2 단백질의 상향조절 및/또는 증가된 세포자멸사 회피의 결과 일 수 있습니다
미토콘드리아는 ATP 생성을 담당하는 소기관이지만 시토크롬 c, 엔도뉴클레아제 G, 세포자멸 유도 인자(AIF)와 같은 많은 세포자살촉진 인자를 포함하고 있어 외부로 방출되면 세포 자살 프로그램을 유도할 수 있습니다.
세포질에서 시토크롬 c의 방출은 세포자멸사 프로테아제 활성화 인자 1(APAF-1) 및 Caspase-9와 연관되어 결국 Caspase-3를 활성화하고 세포자멸사를 실행하는 세포자멸체를 형성합니다
대조적으로, AIF에 의해 시작된 세포자멸사는 카스파제와 무관하며 염색질 응축과 DNA 단편화를 통해 작동합니다
미토콘드리아에서 이러한 pro-apoptotic 단백질의 존재는 암 치료 연구의 흥미로운 표적으로서 세포 소기관에 스포트라이트를 줍니다.
미토콘드리아는 세포자살 유도 과정에서 중심적인 역할을 합니다.
각 미토콘드리아는 세포의 "자가 파괴" 버튼 역할을 한다고 말하는 것이 적절할 것입니다.
암성 미토콘드리아와 비암성 미토콘드리아의 차이는 암세포에서 선택적으로 세포자멸사를 유도하는 세포자멸사 촉진 인자의 방출을 허용하도록 표적화될 수 있습니다.
이 리뷰에서 우리는 미토콘드리아 취약성을 표적으로 하는 치료 조절제 개발의 최근 발전에 대해 논의할 것입니다.
2. 미토콘드리아 취약성 표적화
암세포는 과도한 에너지 요구로 인해 호기성 해당작용 외에 OxPhos에 크게 의존할 수 있습니다
또한, 암세포의 미토콘드리아 막은 정상의 건강한 세포에 비해 과분극되어 잘 조립되어 있지 않습니다
따라서 미토콘드리아 표적 치료법은 정상의 건강한 세포를 선택적으로 보존하면서 암을 특이적으로 표적화할 가능성이 있습니다.
미토콘드리아는 또한 활성 산소종과 산화 스트레스 방어, 에너지 생산 및 세포자멸사 유도에 근본적인 역할을 합니다.
이러한 메커니즘과 기능은 암에서 변경될 수 있으며 치료제가 표적으로 하는 특정 마커 역할을 할 수 있습니다.
암 세포의 필요와 기능의 차이를 이용함으로써 건강한 세포를 보존할 수 있으며 오늘날 많은 화학 요법에서 관찰되는 것처럼 부작용을 피할 수 있는 치료법을 만들 수 있습니다.
예를 들어, Weinberg와 Chandel은 암세포가 단순히 호기성 해당작용을 하는 상태로 변할 수 있지만 미토콘드리아를 표적으로 삼는 것을 지지하는 다른 이유가 있다고 가정합니다
첫 번째는 관류가 잘 되지 않는 종양은 호기성 조건과 포도당에 대한 접근이 제한적일 수 있다는 것입니다. 이것은 암세포가 미토콘드리아 ATP 생성에 의존하도록 하며, 이는 치료법에 의해 구체적으로 표적화될 수 있습니다
두 번째는 일부 암세포가 실제로 ATP 필요량을 OxPhos에 크게 의존한다는 것입니다.
두 시나리오 모두에서 미토콘드리아 표적 치료제는 OxPhos 를 방해하고 암세포를 사멸시킬 수 있습니다.
마지막으로, 미토콘드리아 ATP 생산을 표적으로 하고 억제하는 약물은 암 세포를 호기성 해당 작용 표적 약물에 민감하게 만들고 그 작용을 향상시킬 수 있습니다.
또한, 미토콘드리아가 표적화될 때 pro-apoptotic 요인의 누출은 apoptosis의 활성화를 초래하여 미토콘드리아 표적화 약물 시너지의 개념을 뒷받침합니다.
2.1. 암 치료의 표적으로서의 산화 스트레스 유도
낮은 수준에서 ROS는 증식 및 신호 전달을 촉진하는 데 유리할 수 있습니다
그러나 더 높은 수준에서 ROS는 산화 스트레스를 유도하여 세포 사멸을 유발할 수 있습니다
암세포는 건강한 세포와 달리 증가된 증식 속도를 보완하기 위해 더 높은 농도의 ROS가 필요합니다.
암세포가 결과적으로 증가된 DNA 손상을 이용하여 추가 돌연변이와 종양 형성을 촉진할 수 있다는 가설이 있습니다.
또한, 암세포에 대한 화학요법이나 방사선 요법을 통한 산화 스트레스의 유도는 DNA 손상으로 인한 세포 사멸을 초래할 수 있습니다
대사 변화로 인해 미세 환경에서 증가된 ROS로의 전환은 전이 상태로의 점진적 돌연변이를 허용합니다
결과적으로 암세포는 강화된 산화 환경에서 생존하기 위해 항산화 방어를 상향 조절했습니다.
미토콘드리아에서 ROS의 생성은 OxPhos에서 부산물로 생성되기 때문에 불가피합니다.
ROS의 존재는 미토콘드리아 역학의 변경에 역할 을 하며 결과적으로 수퍼옥사이드 디스뮤타제 및 글루타티온 퍼옥시다제를 포함하는 항산화 방어 시스템을 사용하여 정상 세포에서 효율적으로 소멸됩니다 .
억제되거나 제거되지 않으면 과도한 산화 스트레스는 미토콘드리아 단백질의 기능 장애를 더 일으켜 ROS 생성을 증가시켜 미토콘드리아 손상과 산화 스트레스의 악순환을 생성할 수 있습니다.
이것은 결국 미토콘드리아 막 전위의 붕괴, 막의 투과성 및 세포 사멸의 유도로 이어질 것입니다
2.2. 암 치료를 위한 표적으로서의 미토콘드리아 대사 재프로그래밍
암세포는 필요한 에너지의 대부분을 호기성 해당작용에 크게 의존하고 산소나 포도당 접근이 제한된 상황에서만 OxPhos로 전환합니다.
해당 작용 표적 요법의 사용은 암세포의 증식을 줄이는 데 도움이 될 수 있습니다.
호기성 해당 작용의 상향 조절은 종양 유전자(예: MYC 및 KRAS)의 발현 증가와 P13K 신호 전달 경로의 조절 해제의 결과입니다
과도한 해당과정과 젖산 생성으로 인해 미토콘드리아는 과분극화됩니다.
암세포에 의한 대사 재프로그래밍의 결과로 암세포 대사의 특정 측면은 정상적인 건강한 세포와 구별될 수 있습니다.
이러한 차이는 암 세포에 대한 선택성을 향상시키고 건강한 세포를 표적으로 하는 부작용을 피하기 위한 치료법에 의해 표적이 될 수 있습니다.
미토콘드리아 재프로그래밍 취약성을 표적으로 하는 최근 연구는 암세포에서만 발견되는
상향조절된 대사 단백질의 억제,
미토콘드리아 산화적 인산화 또는 호흡 표적 또는
항생제 유도를 통한 미토콘드리아 기능 장애
등의 다양한 접근 방식에 초점을 맞췄습니다 .
2.3. 미토콘드리아를 표적으로 하여 화학 내성의 민감화 및 역전
암세포 미토콘드리아는 세포자멸사를 피하기 위해 더 많은 양의 항세포자멸사 Bcl2 계열 단백질을 함유하고 있으므로 항암제에 내성이 있습니다.
미토콘드리아 DNA(mtDNA)의 고갈은 생체 내 수많은 암에서 보고되었으며 Bcl2와 같은 항세포사멸 유전자의 발현 증가와 관련이 있습니다.
이것은 생존을 촉진하는 효소를 활성화시켜 화학요법 매개 세포자멸사에 대한 내성을 유발합니다
호기성 해당작용에 의존하기 때문에 암세포는 조절 효소의 상향 조절을 통해 세포 사멸에 저항할 수 있습니다
예를 들어, hexokinase II는 젖산 생산, 세포 증식, 약물 내성 및 침입을 증가시키는 것으로 나타났습니다
젖산 생산은 암 세포가 약간 산성인 미세 환경을 유지하고 젖산염을 항산화제로 사용하는 것과 같은 경로를 통해 생존을 향상시킬 수 있도록 합니다
암세포의 미토콘드리아를 표적으로 하는 화학감작 사례가 많이 보고되었습니다.
한 가지 예는 암세포 미토콘드리아에서 전자 수송 사슬의 복합체 I을 표적으로 하는 약물 메트포르민입니다.
앞서 언급했듯이 내부 정지 난소암 세포는 증식성 세포에 효과적인 화학 요법 치료에 대한 내성을 발달시키므로 암 재발을 예방하기 위해 OxPhos 억제제 요법의 추가가 필요합니다
3. 미토콘드리아를 표적으로 하는 전략
실제로, 미토콘드리아 취약성을 확인하고 암 세포와 비암 세포에서 미토콘드리아 차이를 확인하는 데 광범위한 작업이 투입되었습니다. 이를 통해 암세포 미토콘드리아의 특정 표적화 접근법이 개발되었습니다. 이 섹션에서 우리는 암에 대한 잠재적인 치료제로서 미토칸의 개발로 최근에 이루어진 발전을 요약할 것입니다. Olivas-Aguirre et al. 최근 미토칸에 대한 특정 표적에 초점을 맞춘 T 세포 급성 림프모구 백혈병에 대한 미토콘드리아 표적 요법에 대한 훌륭한 리뷰를 발표했습니다[ 72 ]. 우리는 여러 미토콘드리아 표적을 강조하고 모든 암 유형을 포함하도록 범위를 확장할 것입니다. 우리는 표 1 에서 검토한 모든 치료를 표로 작성 하고 목표를 시각화했습니다
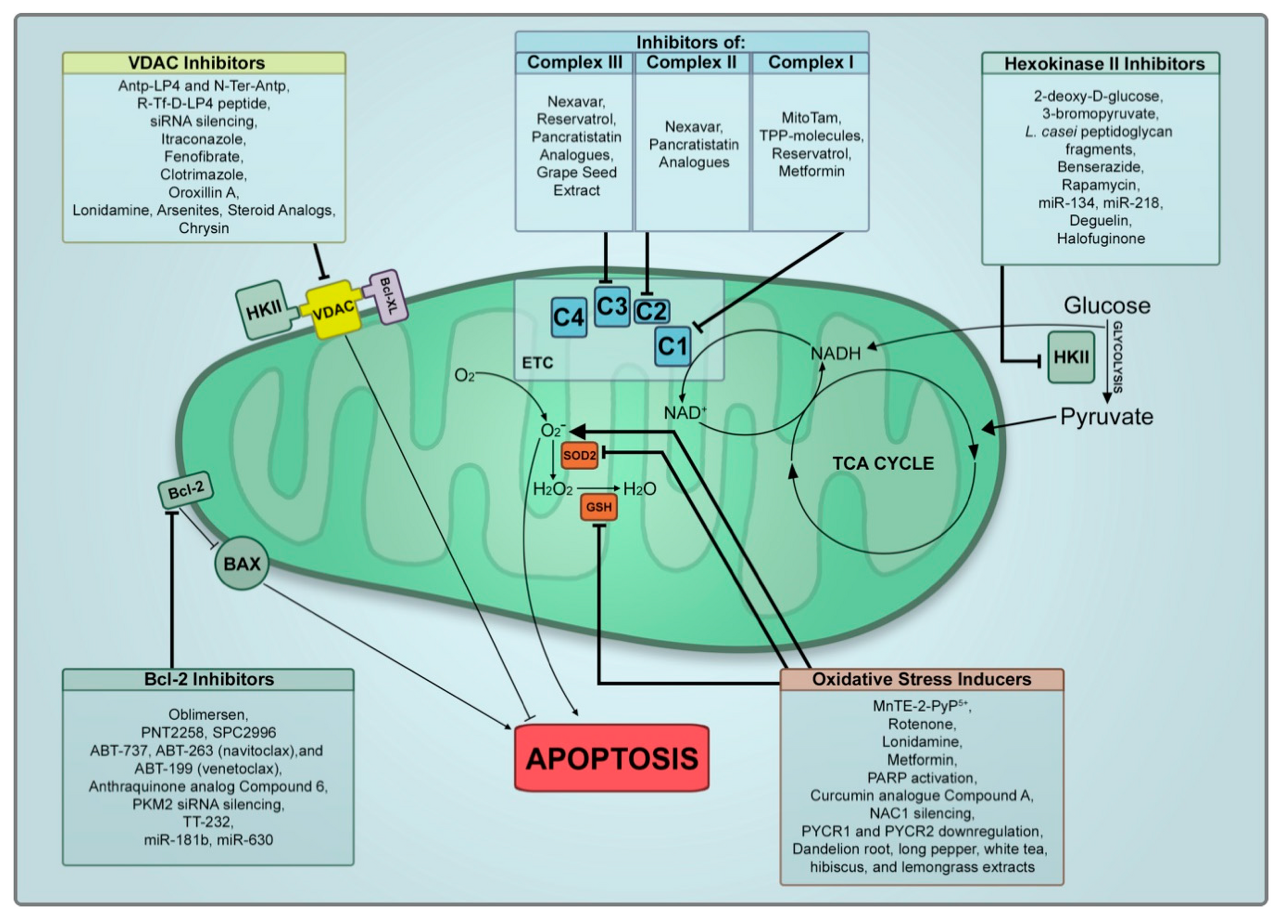
그림 1. 미토칸 치료 및 미토콘드리아 표적. 검토된 미토칸 치료 및 미토콘드리아에서의 표적 요약
HKII, hexokinase II; C1, complex I; C2, complex II; C3, complex III; C4, complex IV; ETC, electron transport chain; VDAC, voltage-dependent anion channel 1; TPP, triphenylphosphonium; PARP, poly (ADP-ribose) polymerase; NAC1, nucleus-accumbens-1; PYCR, pyrroline-5-carboxylate reductase.
표 1. 미토칸 치료, 표적 및 치료 효과.
VDAC1—voltage-dependent anion channel 1; 2-DG—2-deoxy-D-glucose; 3-BPA—3-bromopyruvate; Benz—benserazide; STAT3—signal transducer and activator of transcription 3; HK—hexokinases; PBMCs—peripheral blood mononuclear cells; MMP—mitochondrial membrane potential; PKM2—pyruvate kinase M2 isoform; TPP—triphenylphosphonium; ROS—reactive oxygen species; PARP—poly (ADP-ribose) polymerase; NAC1—nucleus-accumbens-1; PYCR—pyrroline-5-carboxylate reductase; GSE—grape seed extract.
Mitochondrial Targets Mitocan/Treatment Name Treatment Effect Clinical Trial
| Hexokinase II | 2-DG | Cytotoxicity, sensitization to prednisone | NT * | |
| 3-BPA | Cytotoxicity, sensitization to prednisone | NT | ||
| Lactobacillus casei peptidoglycan fragments (European Patent number 1217005) |
Inhibition of entire metabolism of cancer tumour cells |
NT | ||
| Benz | Reduces glucose uptake, lactate production, and ATP levels, led to apoptosis |
Phase 4 | ||
| Rapamycin/siRNA downregulation of STAT3 |
Glycolysis inhibition, reduce glucose consumption |
NT | ||
| miR-134 | Knockdown of HKII reduced glucose consumption leading to apoptosis | NT | ||
| miR-218 | Downregulation of HKII and apoptosis | NT | ||
| VDAC-1 | VDAC1-based peptides Antp-LP4 and N-Ter-Antp |
Highly effective in inducing cell death in leukemia patient PBMCs and cancer cell lines, but not healthy patient PBMCs |
NT | |
| R-Tf-D-LP4 peptide | Targeted transferrin receptor in cancer cells, enhancing specificity of Antp-LP4 and N-Ter-Antp |
NT | ||
| VDAC-1 siRNA silencing | Decreased MMP and ATP levels, reducing tumour burden |
NT | ||
| Itraconazole | Inhibition of cell proliferation | NT | ||
| Fenofibrate | Reprogramming of metabolism and apoptosis in oral carcinomas |
NT | ||
| Clotrimazole | Cytotoxicity, inhibition of glycolysis | NT | ||
| Oroxillin A | Cytotoxicity, apoptosis, cell cycle arrest, and metastasis inhibition |
NT | ||
| Lonidamine | Cytotoxicity | NT | ||
| Arsenites | Cytotoxicity | NT | ||
| Steroid Analogs | Cytotoxicity | NT | ||
| Bcl-2 Family | Oblimersen | Downregulation of Bcl-2, synergy with other treatments |
(G3139) | |
| PNT2258 | Cell cycle arrest, apoptosis in non-Hodgkin’s lymphoma |
Phase 2 | ||
| SPC2996 | Leukemic cell clearance, immune system activation and stimulation |
Phase 2 | ||
| ABT-737 | Apoptosis in lymphoma and leukemia cell lines |
NT | ||
| ABT-263 (navitoclax) and ABT-199 (venetoclax) |
Enhanced effects and specificity compared to ABT-737 | Phase 2 | ||
| Anthraquinone analog Compound 6 | Binds Bcl-2, Mcl-2, and p-Mcl-2 leading to apoptosis induction |
NT | ||
| PKM2 siRNA silencing | Regulates oxidative stress induced apoptosis in a variety of cancers |
NT | ||
| TT-232 | Translocation of PKM2 to nucleus to trigger apoptosis |
Phase 2 | ||
| miR-181b | Sensitize cancer cells to cisplatin | NT | ||
| miR-630 | Sensitize cancer cells to cisplatin | NT | ||
| Electron Transport Chain | Sorafenib (nexavar) | Inhibition of ATP synthase leading to Parkin-mediated apoptosis |
Phase 3 | |
| MitoTam | Increased localization of tamoxifen to mitochondria, leading to increased specificity |
Clinical trials to begin shortly | ||
| TPP-Peptide Artemisinin-TPP Green titania ((G-TiO2-x) conjugated to TPP |
Selectively kill anticancer cells | NT | ||
| Reservatrol | Act as a pro-oxidant leading to cancer cell death |
NT | ||
| Metformin | Selective mitochondrial targeting, acts as an adjuvant with many cancer therapies |
Phase 1 | ||
| Pancratistatin analogues SVTH-6 and SVTH-7 | Highly selective cytotoxicity o n cancer cells in 2D and 3D culture models |
NT | ||
| Oxidative Stress | MnTE-2-PyP5+ | Enhance chemotherapeutic effect by mitochondrial environment modulation |
NT | |
| Rotenone | Activates NOX2 resulting in increased ROS and cell death |
NT | ||
| Lonidamine | Cytotoxicity through ROS generation | NT | ||
| Metformin | Additionally exert oxidative stress | Phase 1 | ||
| PARP activation | Enhances ROS production leading to apoptosis | NT | ||
| Curcumin analogue Compound A | Selective apoptosis through the generation of significant ROS in a variety of cancers |
NT | ||
| NAC1 silencing | Removal of oxidative stress defense mechanism, sensitization |
NT | ||
| PYCR1 and PYCR2 downregulation | Sensitizes cancer cells to ROS by inhibiting stress-response proteins |
NT | ||
| Natural Health Products Targeting Mitochondria |
Chrysin | Inhibits HKII binding to VDAC1 leading to apoptosis |
NT | |
| Deguelin | Downregulates HKII leading to apoptosis | NT | ||
| Halofuginone | Downregulates HKII | Phase 1 | ||
| GSE | Targets complex III and depletes glutathione antioxidant leading to apoptosis in cancer |
NT | ||
| Dandelion root, long pepper, white tea, hibiscus, and lemongrass extracts |
Highly effective induction of apoptosis and excessive ROS generation |
Phase 1 |
3.1. 미토콘드리아 상호작용 헥소키나제 II 표적화
암세포는 호기성 해당작용에 크게 의존하며 결과적으로 포도당 대사의 첫 번째 단계를 촉매하는 헥소키나제(HK)를 과발현합니다
4개의 HK isoform 중에서 hexokinase II(HKII)는 암세포의 생존과 증식에 중요한 역할을 합니다. 외부 미토콘드리아 막에서 HKII는 전압 의존성 음이온 채널 1(VDAC1)에 결합하고 미토콘드리아 내부 막의 아데닌 뉴클레오티드 트랜스로카제와의 상호작용을 촉진합니다
이 상호 작용은 호기성 해당 작용과 OxPhos를 결합할 수 있고 두 대사 과정 사이의 작업 관계를 생성하여 HKII가 미토콘드리아에서 ADP를 ATP로 교환하고 해당 작용 속도를 증가시킬 수 있습니다
HKII를 표적으로 하면 호기성 해당작용의 결합이 해제되고 방해를 받아 암세포가 사망할 수 있습니다.
최근 연구는 HKII를 억제하고 포도당 대사의 진행을 예방할 수 있는 제제에 초점을 맞췄습니다.
중요하게도, 이들 화합물은 건강한 비암성 세포에서 해당작용을 자극하는 동안 암세포에 세포독성을 나타내었으며, 이는 아마도 미토콘드리아 막에 결합하는 HKII에 대한 암세포 의존성 때문일 것입니다.
원래 파킨슨병을 치료하기 위해 고안된 Benserazide(Benz)는 HKII의 또 다른 억제제로 포도당 흡수, 젖산 생산, 세포 내 ATP 수준을 선택적으로 감소시켜 미토콘드리아 막 전위(MMP)와 세포자멸사를 소산시킬 수 있습니다
HKII를 유전적으로 표적화하여 암세포를 죽이려는 노력도 있었습니다.
신호 변환기 및 전사 활성제 3(STAT3)은 종양 발달, 혈관신생 및 전이에서 중요한 역할을 하는 종양유전자입니다
STAT3는 라파마이신(mTOR)의 하류 인자이자 HKII의 조절 인자로 알려져 있습니다.
따라서 mTOR-STAT3-HKII 경로는 암세포에서 해당 작용 억제에 대한 흥미로운 표적입니다
라파마이신 처리와 STAT3의 siRNA 하향 조절은 포도당 소비를 직접적으로 감소시키고 HKII를 하향 조절하는 것으로 나타났습니다
HKII의 녹다운은 microRNA-143(miR-143) 과발현을 사용하여 달성되었으며, 포도당 대사 및 증식 억제를 통해 암세포의 세포자멸사를 촉진했습니다
같은 맥락에서, miR-218의 과발현은 새로운 miR-218/Bmi1/HKII 축에서 세포 촉진 발암 유전자 Bmi1에 의해 촉진되는 HKII 발현을 하향 조절했습니다
흥미롭게도 HKII는 암 세포를 화학 요법과 방사선 요법에 민감하게 만드는 표적으로 연루되어 있습니다. HKII 고갈은 또한 암세포가 OxPhos에 의존하게 하여 암세포를 감작시켜 복합 I 억제제 메트포르민의 표적이 되도록 하여 세포 사멸을 촉진합니다
다른 연구에서는 HKII와 그 조절자를 모두 표적으로 삼는 것이 방사선 치료에 감작화되는 것으로 나타났습니다
3.2. VDAC1 표적화
VDAC1(Voltage-dependent anion channel 1)은 미토콘드리아 외막에 위치하며 대사산물, 지방산, Ca 2+ , ROS 및 콜레스테롤을 포함한 많은 화합물 이 미토콘드리아 막을 가로질러 이동할 수 있도록 합니다
VDAC1을 표적으로 함으로써 암세포의 세포자멸사 회피 메커니즘을 방해할 수 있습니다.
VDAC1은 미토콘드리아 세포사멸에서 중심적인 역할을 하며, Bcl2 또는 Bcl-xL과 같은 항-세포사멸 단백질 및 세포사멸 억제 HKII와 상호작용합니다.
VDAC1 결합 부위에 기반한 합성 펩타이드는 Bcl2, Bcl-xL 및 HKII를 억제하여 VDAC1과의 결합을 방지하여 암세포가 세포 사멸을 회피하는 능력을 억제하는 데 사용되었습니다
이 단백질을 표적으로 하기 위한 유전적 접근을 취함으로써, VDAC1의 siRNA 침묵은 암 세포주에서 감소된 MMP 및 ATP 수준뿐만 아니라 폐암 이종이식된 마우스에 대한 종양 부담의 급격한 감소를 초래했습니다
항진균제인 이트라코나졸은 VDAC1을 표적으로 하고 미토콘드리아 대사의 조절을 통해 mTOR 활성과 세포 증식을 억제하여 세포자멸사를 유도하는 것으로 나타났습니다
그러나 VDAC1 녹다운은 건강한 세포에서 발현되기 때문에 독성 영향을 미칠 수 있으며 독성에 대한 더 많은 연구가 수행되어야 한다
또한, VDAC1-HKII 복합체는 HKII를 과발현하는 암세포에서 세포자멸사를 유발하도록 표적화될 수 있습니다.
최근 연구에서 fenofibrate는 HKII와 VDAC1의 결합을 차단하고 구강 편평 세포 암종의 대사 경로를 재프로그래밍했습니다
VDAC-HKII 결합을 파괴할 수 있는 다른 많은 화합물은 클로트리마졸, 3-BR, 2DG, 오록실린 A, 로니다민, 아세나이트 및 스테로이드 유사체를 포함합니다.
페노피브레이트(임상시험번호: NCT01965834)와 로니다민(임상시험번호: NCT00435448)은 임상에 들어갔으나 각각 2상과 3상에서 종료됐다.
3.3. Bcl2 타겟팅
항-아폽토시스 B 세포 림프종 2(Bcl-2) 단백질 계열은 세포 사멸의 세포 생존 억제를 촉진하는 역할 때문에 암 치료에 흥미로운 표적을 제시합니다
Bcl-2를 표적으로 하는 약제를 개발하려는 초기 시도는 RNA 안티센스 분자의 전달을 통해 Bcl-2 수준을 감소시키는 것을 목표로 했습니다
3.4. 전자 수송 사슬 복합체 표적화
암세포 OxPhos를 표적으로 하도록 설계된 치료법은 암세포 대사에서 중요한 기계를 차단하여 호기성 해당작용 표적 제제에 민감하게 하거나 잠재적으로 미토콘드리아 pro-apoptotic protein의 누출을 촉진할 수 있습니다.
복합체 I, II 및 III를 표적으로 하는 많은 미토칸은 암에 대해 큰 효능과 선택성을 보여주었습니다.
Sorafenib(nexavar)은 ATP 합성효소와 함께 ETC의 complex II 및 complex III의 활성을 억제하여 serine-threonine protein kinase PINK1의 안정화를 유도하고, 이는 차례로 Parkin 매개 세포자멸사를 유도합니다
아르테미시닌 및 녹색 티타니아(G-TiO)와 같은 다른 미토칸2-x 는 TPP에 접합하여 암세포를 선택적으로 죽이는 것으로 나타났습니다.
Reservatrol은 복합체 I 및 III에 작용 및 억제하고 산화촉진제로 작용하여 암에서 세포 사멸을 유도하는 것으로 나타났습니다
메트포르민은 ETC 복합체를 표적으로 하는 것이 암에 대한 효과적인 단일 또는 보조 치료를 가능하게 한다는 확실한 증거를 제공합니다.
3.5. 산화 스트레스 타겟팅
미토콘드리아 OxPhos가 표적이 되면 미토콘드리아 스트레스가 ROS 생성을 증가시켜 산화 스트레스를 유발할 수 있습니다.산화촉진제의 사용은 암세포의 ROS를 세포독성 수준으로 증가시켜 세포자멸사를 유도합니다.
암세포는 증식 신호, 추가 종양 형성에 대한 돌연변이 비율 증가, DNA 손상 및 게놈 불안정성을 위해 ROS의 존재 하에서 번성합니다
따라서 암세포는 이미 정상의 건강한 세포에서 발견되지 않는 선택적 취약성을 나타내는 더 높지만 치명적이지 않은 산화 스트레스를 가질 수 있습니다.
(PARP)의 활성화는 PARP-1-ATF4-MKP-1-JNK/p38-MAPK 역행 경로를 향상시켜 ROS 생성에 이어 세포 사멸을 유도합니다
우리는 또한 새로운 curcumin 유사체인 Compound A가 다양한 암 세포주에서 단독으로 그리고 또 다른 산화촉진제인 piperlongumine과 조합하여 ROS의 생성을 통해 높은 효능을 나타내고 선택적 세포자멸사를 유도한다는 것을 입증했습니다
대조적으로, 암세포의 산화 스트레스 방어 기전과 관련된 단백질의 억제는 표적이 될 수 있습니다.
그렇게 함으로써 암세포는 정상적인 건강한 세포에 비해 더 높은 농도의 ROS를 포함하는 환경에서 번성하는 능력을 잃을 수 있습니다.
그러나 이러한 산화 스트레스 표적화 방법은 건강한 비암성 세포에서 ROS 보호 메커니즘의 필요성과 높은 발현 때문에 이상적이지 않을 수 있습니다.
Piperlongumine은 또한 산화 스트레스 보호 메커니즘을 억제하여 암세포를 사멸시키는 글루타티온 S-트랜스퍼라제 PI(GSTP1)의 표적화와 관련이 있습니다
3.6. 여러 취약점을 대상으로 하는 복합 요법
암세포 취약성의 여러 측면을 표적으로 삼으면 여러 환경에서 암세포를 표적으로 삼아 암의 재발을 추가로 예방할 수 있습니다. 예를 들어, 호기성 해당작용과 OxPhos를 모두 표적으로 하면 암세포가 세포자살을 일으킬 수 있습니다.
PST 유사체 SVTH-7에 대한 우리의 연구는 산화 스트레스 유도 piperlongumine(PL)과의 조합 치료가 MMP를 소산시키고, 산소 소비를 감소시키며, 미토콘드리아에서 많은 세포자멸사 인자의 방출을 유도하는 보다 효과적인 치료로 이어짐을 나타냈습니다
결론
미토콘드리아는 세포자멸사, OxPhos를 통한 ATP 생성 및 특히 미토콘드리아 기능 장애의 경우 ROS의 원천으로서 중심입니다.
이것은 세포 사멸 유도를 위해 암세포를 표적으로 하는 매우 흥미로운 세포 소기관을 제시합니다.
암세포와 건강한 세포 미토콘드리아의 차이 때문에 미토콘드리아를 표적으로 삼는 것은 DNA 복제 및 복구를 표적으로 삼는 것보다 훨씬 더 선택적인 것입니다. 선택적 치료를 사용함으로써 접근법은 정상적인 건강한 세포에 대한 세포 독성으로 인해 발생하는 장기 독성과 같은 부작용을 피할 수 있습니다.
미토콘드리아 표적 약물은 암세포의 미토콘드리아 취약성을 표적으로 삼을 수 있지만 건강한 세포는 표적으로 삼을 수 없습니다.
다른 치료법과 함께 보조제로서 미토칸을 사용하면 암 약물 내성을 억제하여 암세포를 효율적으로 제거할 수 있습니다.
암 세포의 미토콘드리아 차이와 결합된 산화 스트레스 취약성은 미토칸을 산화 촉진 조절제와 함께 사용하여 암세포를 죽일 수 있는 훌륭한 기회를 제공합니다.
'Ferroptosis' 카테고리의 다른 글
| ferroptosis의 바이오 마커 및 기여자로서의 ACSL4 (0) | 2023.03.22 |
|---|---|
| 죽음이 우리를 갈라놓을 때까지: 자가포식과 세포자멸사의 결혼 (0) | 2021.11.20 |
| 표적으로서의 VDAC1: 세포자멸사에서의 역할에 초점 (0) | 2021.11.19 |
| 암 화학 요법을 보완하는 천연물질: 자가포식-아폽토시스 경로의 약리학적 조절 (0) | 2021.11.19 |
| ER 스트레스 매개 세포자멸사를 유도하는 항암 천연물 (0) | 2021.11.10 |