2020
Lipid metabolism and cancer | Journal of Experimental Medicine | Rockefeller University Press
SREBP activation is regulated by sterol fluctuations in the ER. Cholesterol binds to SCAP, thereby disrupting the interaction between SCAP and COPII and retaining SREBP in the ER (Shimano and Sato, 2017). Oxysterols, such as 25-hydroxycholesterol, are much
rupress.org
Lipid metabolism and cancer
지질 대사의 조절 장애는 암에서 가장 두드러진 대사 변화 중 하나입니다.
암세포는 지질 대사를 이용하여 에너지, 생물학적 막의 구성 요소, 증식, 생존, 침습, 전이 및 종양 미세 환경 영향 및 암 치료에 대한 반응에 필요한 신호 분자를 얻습니다.
지질은 단백질 및 핵산과 함께 세포를 구성하는 생물학적 막 및 빌딩 블록의 필수 구성 요소입니다.
또한 지질은 에너지 저장 및 대사에 사용되며 많은 세포 활동에 대한 신호 분자로서 중요한 역할을 합니다.
지질 흡수, 합성 및 가수분해와 같은 지질 대사의 조절은 세포 항상성의 유지에 필수적입니다.
종양이 진행되는 동안 영양소 가용성이 지속적으로 변화하는 종양 미세 환경의 암세포는 지질 대사를 활용하여 빠른 증식, 생존, 이동, 침습 및 전이를 지원합니다.
당지질과 인지질(포스포글리세리드와 스핑고리피드로 세분화됨)은 콜레스테롤과 함께 생체막의 주요 구성 요소입니다.
콜레스테롤은 또한 지용성 비타민과 스테로이드 호르몬 합성을 위한 기질입니다.
당지질과 인지질의 주요 성분인 지방산(FA)은 글리세롤 부분과 함께 에스테르화되어 트리글리세리드를 형성할 수 있습니다.
트리글리세리드는 비극성 지질로서 충분한 영양 가용성 동안 지질 방울에 합성 및 저장되고 에너지 스트레스 조건에서 FA 산화(FAO, β-산화라고도 함)에 의해 ATP를 생성하기 위해 가수분해됩니다.
에너지 대사 및 막 형성 외에도 지질은 막 지질의 포스포리파제 의존성 가수분해 및 필수 FA 합성에서 파생된 2차 메신저를 형성하며, 이의 이용 가능성은 주로 식단의 지질에 의해 결정됩니다.
종양 형성을 촉진할 수 있는 기타 여러 신호 축의 활성화를 유발합니다.
또한, 옥시스테롤 및 콜레스테롤을 포함한 스테롤은 다운스트림 유전자 발현을 위한 스테롤 조절 요소(SRE)-결합 단백질(SREBP) 활성화의 중요한 조절자이며, 따라서 이들 수준은 암의 지방 생성에 영향을 미칩니다.
콜레스테롤은 신호 전달을 위한 지질 뗏목의 구성 요소이며 Hedgehog 신호 활성화를 위해 Hedgehog 및 Smoothened 단백질을 공유 결합으로 수정할 수도 있습니다.
지질 흡수
FA 활용
포유류는 특정 FA, 즉 탄화수소 사슬의 Δ9 위치에 이중 결합을 갖는 FA만 생성합니다.
다른 FA, 특히 다중불포화 FA는 필수적이며 식단에서 얻습니다.
원형질막에서 알려진 FA 단백질 수송체에는 분화 클러스터 36(CD36, FA 트랜스로카제라고도 함), FA 수송 단백질 패밀리(총칭하여 SLC27이라고도 함) 및 원형질막 FA-결합 단백질(FABP)이 포함됩니다.
모두 종양에서 증가된 유전자 및 단백질 발현을 나타냅니다.
불포화 지방산을 제공하기 위해 외인성 지질을 사용하는 암세포의 능력은 산소 수준과 발현되는 종양 유전자 유형에 따라 달라지며 모든 지질이 동등하게 활용되는 것은 아닙니다.
저산소성 종양 세포는 저산소증 유도 인자-1α(HIF-1α)로 유도된 FABP3/7 발현에 따라 증가된 FA 수입을 나타내며, 이는 포도당의 아세틸-코엔자임 A(CoA)로의 이화작용이 감소하기 때문에 감소된 새로운 FA 합성을 동반합니다.
혈액 자원이 되는 것 외에도 종양 미세 환경의 지방 세포는 종양 세포에 세포외 FA를 제공합니다. .
Omentum-전이성 난소 세포는 지방 세포에서 지방 분해를 활성화하여 FAO 증가 및 빠른 종양 성장을 위해 FABP4 의존적 방식으로 암세포에 의해 후속적으로 분비 및 흡수될 수 있는 유리 FA를 생성합니다.
따라서 저산소증, 종양 세포에서 과발현된 FA 수송체, 특정 암유전자 발현, 지방세포 및 섬유아세포를 포함한 종양 세포 조절 기질 세포는 종양 세포가 세포외 FA를 흡수하고 종양 세포 증식을 유지하기 위한 유사분열 신호를 생성하도록 유도할 수 있습니다.
콜레스테롤 흡수
식이 콜레스테롤은 Niemann-Pick type C1-like 1(NPC1L1) 단백질이 장 장세포막에 흡수되며, 여기서 콜레스테롤은 간에서 흡수를 위해 acylCoA:cholesterol acyltransferases(ACATs, sterol O-라고도 함)에 의해 에스테르화됩니다.
주요 콜레스테롤 생합성 기관인 간은 콜레스테롤을 혈류에 초저밀도 지단백질로 전달하고, 여기서 초저밀도 지단백질은 LDL 수용체(LDLR)에 의해 흡수되도록 저밀도 지단백질(LDL)로 처리됩니다.
지방 생성
정상 조직의 지방 생성은 주로 간세포와 지방세포로 제한됩니다. 그럼에도 불구하고 암세포는 외인성 지질 공급원이 있는 경우에도 높은 대사 요구 또는 종양 미세 환경에서 혈청 유래 지질 결핍에 대한 반응으로 지방 생성을 활성화합니다( Röhrig and Schulze, 2016 ). 지질 합성의 주요 기질은 세포질 아세틸-CoA이며, 이는 ATP-시트레이트 리아제(ACLY)에 의한 시트레이트 또는 아세틸-CoA 합성 효소에 의한 아세테이트(ACSS; Li et al., 2017a )에 의해 유도될 수 있습니다. 포도당과 글루타민의 탄소는 구연산염 생산에 기여합니다( Li et al., 2016b ; Metallo et al., 2011 ; 그림 1).
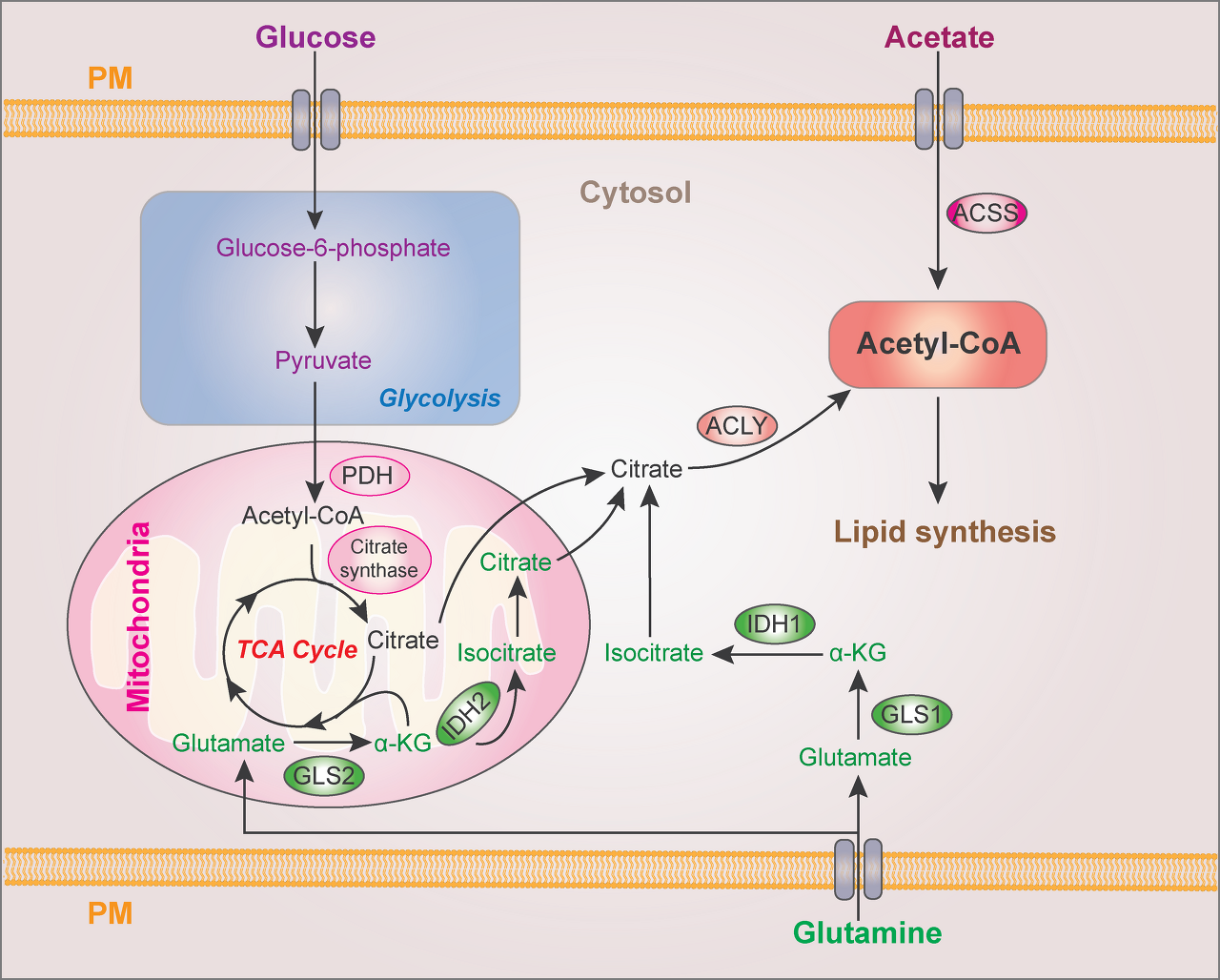
그림 1. 지질 합성을 위한 세포질 아세틸-CoA 생산.
세포질 아세틸-CoA는 ACLY 촉매 구연산염과 ACSS 촉매 아세테이트에서 생산됩니다.
포도당과 글루타민은 각각 TCA 회로에서 미토콘드리아 피루브산 산화와 환원성 카르복실화로부터 구연산염 생산에 기여합니다.
PDH, pyruvate dehydrogenase; α-KG, α-ketoglutarate; GLS, glutaminase; PM, plasma membrane.
포도당 유래 피루브산은 피루브산 탈수소효소에 의해 아세틸-CoA로 전환되고, 이어서 구연산 합성효소가 매개하는 구연산 생산을 통해 미토콘드리아 구연산 수송 단백질에 의해 미토콘드리아에서 세포질로 배출됩니다.
글루타민은 세포질 글루타미나아제 1(GLS1) 또는 미토콘드리아 GLS2에 의해 매개되는 α-케토글루타레이트로 전환되고, 이어서 세포질 이소시트레이트 탈수소효소 1(IDH1) 및 미토콘드리아 IDH2 의존성 이소시트레이트 및 후속 시트르산 생성이 뒤따릅니다.
FA와 콜레스테롤은 모두 일련의 반응을 통해 아세틸-CoA에서 합성되기 때문에 아세틸-CoA 수준은 지질 생산의 핵심 요소입니다( 그림 2 ).
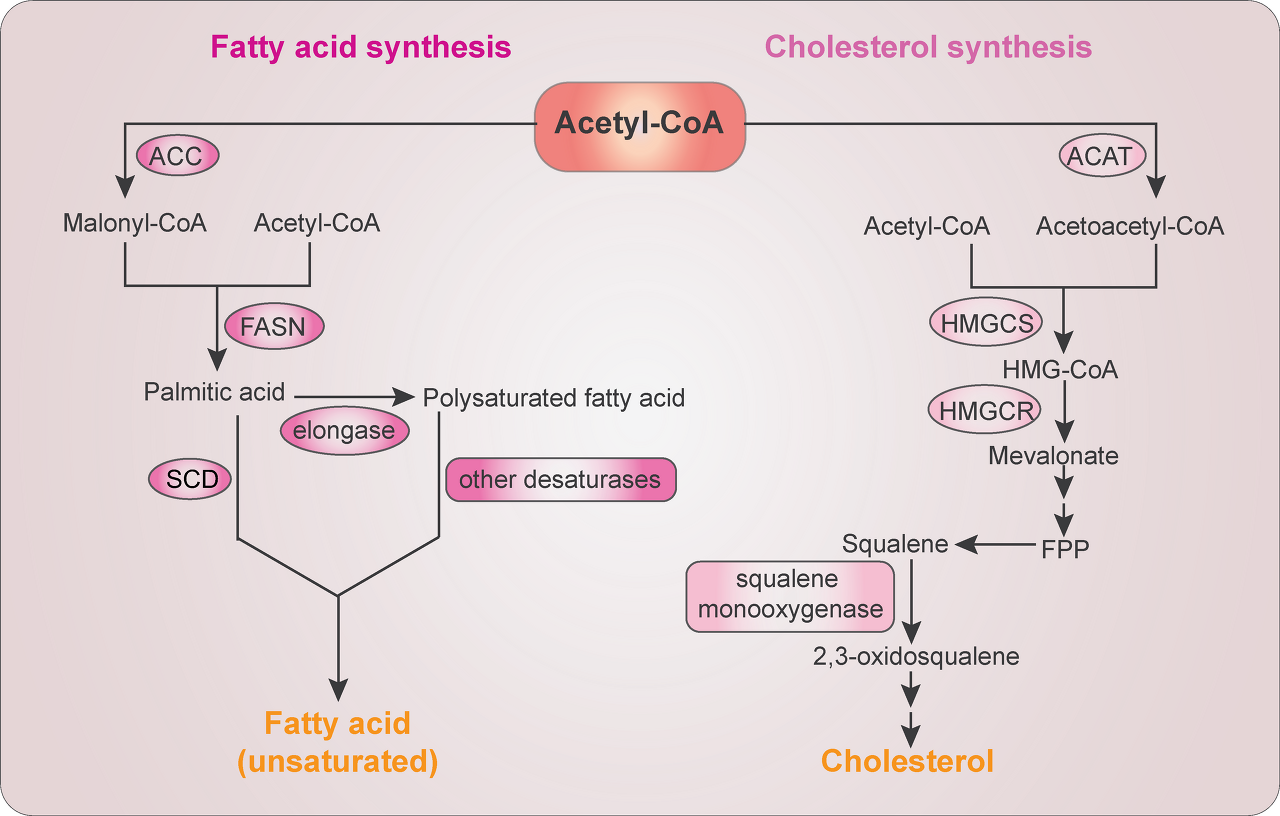
FA와 콜레스테롤 생합성.
FA 생합성은 ACC에 의해 아세틸-CoA가 말로닐-CoA로 전환되는 것으로 시작됩니다.
Acetyl-CoA 및 malonyl-CoA는 FASN에 의해 팔미트산으로 촉매화됩니다.
추가 elongation은 elongase에 의해 매개되어 다중포화 FA를 형성합니다.
팔미트산 및 다중포화 FA는 각각 SCD 및 기타 지방 아실-CoA 불포화 효소에 의해 불포화 FA로 탈포화됩니다.
콜레스테롤 생합성은 ACAT에 의해 두 분자의 아세틸-CoA가 축합되어 아세토아세틸-CoA를 형성하는 것으로 시작되며, 이는 HMG-CoA 합성효소(HMGCS)에 의해 세 번째 아세틸-CoA 분자와 추가로 축합되어 HMG-CoA를 형성합니다.
그런 다음 HMGCR은 HMG-CoA를 메발로네이트로 환원시키고 이는 파르네실 피로포스페이트(FPP)로 전환됩니다.
파르네실 피로인산으로 전환된 스쿠알렌은 SM에 의해 산화되어 콜레스테롤과 스테롤의 전구체인 2,3-옥시도스쿠알렌을 생성합니다.
Acetyl-CoA –생성 효소 ACLY 및 ACSS
ACLY
ACLY는 시트레이트와 CoA를 옥살로아세테이트와 아세틸-CoA로 전환하는 것을 촉매합니다.
ACLY를 억제함으로써 bempedoic acid는 환자의 LDL 콜레스테롤 수치를 감소시켜, 지방 생성에서 ACLY의 중요한 역할을 나타냅니다.
감소된 ACLY 발현은 종양 세포 생존력을 감소시키고 다양한 암 유형에서 종양 세포 증식, 침습 및 전이를 억제합니다.
SREBP1에 의해 전사적으로 상향 조절되는 ACLY는 해당 중간체 과당 6-인산에 의해 알로스테릭하게 활성화되고 ALCY의 동종 알로스테릭 조절을 통해 고농도의 시트르산에 의해 억제됩니다.
세포질에서 지질 대사에 참여하는 것 외에도 핵으로 전위된 ACLY는 히스톤 아세틸화 및 유전자 전사 조절을 위해 아세틸-CoA를 생성합니다.
신경교종 및 전립선암 세포에서 성장 인자 또는 발암성 K-Ras 발현은 AKT-ACLY 신호 전달, 히스톤 아세틸화, 세포 증식 및 종양 성장을 촉진합니다.
따라서 ACLY 매개 아세틸-CoA 생산은 세포질과 핵 모두에서 여러 세포 활동에 중요한 역할을 합니다.
ACSS2
ACSS는 아세테이트와 CoA의 연결을 통해 아세틸-CoA를 생성합니다.
ACSS1과 ACSS3은 미토콘드리아 단백질이고 ACSS2는 세포질과 핵에 국한되어 있습니다.
SREBP에 의해 전사적으로 상향 조절되는 ACSS2는 인간 종양의 많은 부분에서 발현되며 특히 대사 스트레스 하에서 암세포 성장을 유지하기 위한 아세테이트 촉매 작용에 중요합니다.
아세테이트는 장내 미생물총에 의해 과당에서 전환될 수 있으며, 미생물총의 고갈 또는 간 ACSS2의 침묵은 볼루스 과당의 간 아세틸-CoA 및 FA로의 전환을 억제합니다.
또한 ACSS2 고갈 또는 결핍은 종양 세포 성장 및 마우스 종양 형성을 크게 억제하여 종양 성장을 위한 지질 바이오매스 생산에서 아세테이트 소비의 중요한 역할을 강조합니다.
제한된 양의 산소, 혈청 또는 포도당은 ACSS2의 핵 국소화를 증가시킵니다.
포도당 결핍은 5' AMP 활성화 단백질 키나제(AMPK) 매개 ACSS2 S659 인산화를 촉진하여 교모세포종 세포에서 핵 전위를 위한 핵 국소화 신호를 노출시킵니다.
또한 ACSS2는 간 X 수용체(LXR)/레티노이드 X 수용체 전사 인자의 발현 및 활성화를 향상시켜 장기간의 단식 동안 지방 생성을 촉진합니다.
인간 종양 표본의 분석에 따르면 ACSS2 S659 인산화는 신경교종 악성종양 및 비-SCLC 환자의 낮은 생존율과 양의 상관관계가 있는 반면(NSCLC) 높은 ACSS1/2 발현은 HCC 환자에서 히스톤 H3 아세틸화 및 FA 합성효소(FASN) 발현을 증가시켰습니다.
이러한 발견은 새로운 지방 생성과 리소좀 생물 생성, 자가 포식 및 지질 대사를 촉진하기 위한 유전자 발현의 후성 유전적 조절의 직접적인 관련에서 ACSS2의 이중 역할을 보여줍니다.
FA 생합성 효소
아세틸-CoA 카르복실라제(ACC)
FA 합성을 위한 속도 제한 효소인 ACC는 아세틸-CoA에서 말로닐-CoA로의 카르복실화를 촉매합니다( 그림 2 ; Wei and Tong, 2015 ).
포유동물 ACC에는 2개의 조직 특이적 동형 이 있습니다.
ACC1은 주로 지방 생성 조직(간 및 지방 조직)에서 발현되는 세포질 효소이며 FA 합성에 중요합니다.
대조적으로, ACC2는 미토콘드리아 외막에 내장되어 주로 산화 조직(심장 및 골격근)에서 발현되며, ACC2 생성 말로닐-CoA는 카르니틴 팔미토일트랜스퍼라제 I(CPT1, 팔미토일-CoA 트랜스퍼라제 I, CAT1 또는CCAT이라고도 함)을 억제합니다.
미토콘드리아에 의한 FA 흡수 및 FAO의 속도 제한 단계를 제어합니다.
ACC1 결핍은 FA 합성을 감소시키고 전립선 및 유방 종양 세포의 세포자멸사를 유도하지만 비악성 세포는 유도하지 않습니다.
ACC1은 전사 수준에서 SREBP에 의해 조절되고 단백질 수준에서는 인산화, 알로스테릭 조절자의 결합, 단백질-단백질 상호작용의 복잡한 상호작용에 의해 조절됩니다.
AMPK는 ACC1 S79 및 ACC2 S212를 인산화하여 활성을 억제하는 반면, ACC1/2 인산화--dead mutants의 발현은 마우스 간의 지방 생성 및 병변 및 인간 간암 세포의 증식을 증가시킨다.
ACC2 발현 수준은 임상 암 단계 및 감소된 5년 생존율과 양의 상관관계가 있다.
영양소가 풍부한 동안 프롤릴 하이드록실라제 도메인 단백질 3(PHD3)은 ACC2를 수산화하고 활성화하여 FAO를 억제합니다.
급성 골수성 백혈병(AML)에서 PHD3 발현은 극적으로 감소하여 AML 세포 증식 및 질병 중증도를 유발하는 FAO의 증가에 기여합니다.
FASN
FASN 과발현 및 과다 활동은 많은 인간 상피암 및 이들의 전종양 병변에서 일반적으로 발생하며 암 재발 및 사망의 더 높은 위험과 상관관계가 있습니다.
FASN 발현은 PTEN 손실, 스테로이드 호르몬, PI3K-AKT 및 ERK1/2 신호 전달 캐스케이드의 활성화에 의존하는 방식으로 EGFR 및 ERBB2의 활성화에 의해 자극되는 것으로 나타났습니다.
종양 관련 FASN 과발현은 SREBP1에 의해 전사 수준에서 우선적으로 조절됩니다
FASN 억제는 FA 합성을 감소시키고 말로닐-CoA 축적을 유도하여 CPT1 매개 FAO를 억제하고 후속적인 세포 주기 정지 및 종양 세포의 세포자멸사를 유발합니다.
FASN 억제는 막 인지질 구성, 구조, 유동성 및 지질 뗏목 형성을 변경하여 EGFR 및 ERBB2와 같은 티로신 키나제 수용체의 정확한 위치 및/또는 기능을 손상시킵니다.
FASN 의 제거는 마우스 간세포암 형성을 상당히 지연시켰습니다.
특히, FASN 결핍은 콜레스테롤 생성을 위한 핵 전위 및 SREBP2의 활성화를 촉진했습니다.
HCC 발달에서 FASN 의존성 FA 합성 및 SREBP2 매개 콜레스테롤 생성의 중요한 역할을 강조합니다.
Stearoyl-CoA desaturase=스테아로일-CoA 불포화효소(SCD)
SCD가 기능하려면 NADPH와 산소가 필요하며, 저산소 조건에서 암세포가 불포화 FA 함유 지질의 외인성 공급에 더 의존하는 이유를 추가로 설명합니다.
세포에 외인성 지질이 결핍되면 후속 SCD1 억제가 페로프토시스와 세포자멸사를 모두 유도했습니다.
SCD1의 억제는 내인성 막 항산화제인 CoQ10을 감소시킵니다.
CoQ10의 고갈은 ferroptosis와 관련이 있습니다.
ER 막을 포함한 막의 인지질에서 불포화 지방 아실 사슬의 동시 감소 및 장쇄 포화 세라마이드의 증가는 펼쳐진 단백질 반응, ER 스트레스 및 세포자멸사를 유도합니다.
억제된 불포화는 미토콘드리아 호흡을 손상시켜 산화 스트레스를 유발하고 MUFA가 카디오리핀(사이토크롬 c에 결합하여 막으로부터의 방출을 막는 미토콘드리아 내부 막에만 있는 특정 종류의 지질)으로 통합되는 것을 감소시킵니다.
SCD 억제는 증가된 시토크롬 c 방출 및 세포자멸사와 일치합니다.
SCD1의 억제는 간 종양 시작 세포가 ER 스트레스에 의해 유도된 펼쳐진 단백질 반응을 통해 분화하도록 하여 소라페닙에 대한 감수성을 향상시킵니다.
이종이식 마우스 모델에서 SCD1 침묵 또는 억제는 또한 인간의 위, 결장, 폐 및 전립선암 세포에서 유래한 종양의 형성을 감소시켰으며, 이는 종양 발달에서 SCD1의 중요한 역할을 강조합니다.
콜레스테롤 생합성 효소
Mammalian 3-hydroxy-3-methylglutaryl (HMG)–CoA reductase (HMGCR)
콜레스테롤 생합성을 위한 메발로네이트 경로의 속도 제한 효소인 HMGCR은 ER에 국한된 당단백질이며 HMG-CoA를 메발로네이트로 전환합니다( 그림 2 ; Liscum et al., 1985 ).
HMGCR의 조절은 전사, 전사 후, 번역 및 번역 후 수준에서 달성됩니다.
HMGCR 유전자 전사는 SREBP2에 의해 활성화되는 반면, mRNA 번역은 알려지지 않은 메발로네이트 유래 비스테롤 대사산물에 의해 차단될 수 있습니다.
HMGCR 분해는 스테롤, 주로 옥시스테롤 및 메틸화 스테롤, 및 비타민 E 계열 구성원인 δ-토코트리에놀 및 γ-토코트리에놀에 의해 유도될 수 있습니다. .
저산소 상태에서 저산소증 유도 인자-1α는 INSIG2 전사를 활성화하여 HMGCR 분해를 촉발하고 간에서 콜레스테롤 생합성을 억제합니다.
AMPK는 HMGCR S872를 인산화하고 HMGCR 의존성 콜레스테롤 생합성을 억제합니다
HMGCR 발현은 위암, 교모세포종 및 전립선암을 비롯한 여러 유형의 암에서 상향 조절됩니다.
HMGCR의 과발현은 이러한 암세포의 성장과 이동을 촉진하는 반면 HMGCR 녹다운은 종양 형성을 억제합니다.
HMGCR 억제는 고형암 및 혈액암 및 약물 내성이 있는 암을 치료하기 위한 표적이 되었습니다.
흥미롭게도, 친유성 HMGCR 억제제 심바스타틴 치료는 단백질 프레닐화에 필요한 메발로네이트 경로 생성 게라닐게라닐 이인산과 항원 제시 세포에서 작은 구아노신 트리포스파타제 Rab5의 프레닐화 수준을 감소시켜 엔도솜 성숙을 정지시키고 항원 보유 기간을 연장하고, 항원 제시 및 항원 특이적 항종양 면역. 또한 심바스타틴 치료는 암 예방 접종을 강력하게 강화하고 종양 치료를 위한 항 PD-1 항체와 상승 작용을 합니다.
이러한 결과는 암 면역 요법을 강화하기 위한 표적으로서 메발로네이트 경로의 잠재력을 강조합니다.
Squalene monooxygenase (SM)
HMGCR 의 하류에 있는 두 번째 속도 제한 ER 관련 콜레스테롤 생합성 효소인 SM( SQLE 로 인코딩됨)은 nonsterol intermediate squalene을 2,3(S)-oxidosqualene으로 전환합니다( 그림 2 ).
HMGCR과 유사하게 SM은 SREBP2의 표적이기도 합니다.
콜레스테롤과 달리 올레이트와 같은 불포화 FA는 MARCH6 매개 분해를 억제하여 SM을 안정화 시켜 콜레스테롤 합성을 유지 합니다.
SQLE 유전자좌는 여러 암에서 복제수를 증가시켰고 , 비알코올성 지방간 질환으로 유발된 간세포암종에서 과발현이 검출되었으며, 췌장암의 방사선 내성 및 유방암, 전립선암, 결장직장암, 편평 폐암의 진행 또는 불량 예후와 관련이 있음 ( Brown et al., 2016 ; Cirmena et al., 2018 ; Liu et al., 2018 ). 또한 SCLC 세포의 증식은 콜레스테롤 생합성 억제가 아니라 SM 기질 스쿠알렌의 독성 축적으로 인한 SM 억제에 민감합니다( Mahoney et al., 2019). 역형성 림프종 키나제-양성 역형성 대세포 림프종 세포에서 발견된 SM 발현의 손실은 콜레스테롤 영양요구성에 기여하고 이러한 세포가 콜레스테롤 흡수 및 종양 성장을 위해 LDLR에 의존하게 만듭니다( Garcia-Bermudez et al., 2019 ). 이러한 발견은 다양한 유형의 종양에서 콜레스테롤 대사의 뚜렷한 특징을 나타냅니다.
지방 생성의 전사 조절
지방 생성은 SREBF1 유전자에 의해 암호화되는 SREBP1a 및 SREBP1c, 그리고 SREBF2 유전자에 의해 암호화되는 SREBP2 의 세 가지 이소형으로 구성된 나선-루프-나선 류신 지퍼 전사 인자 계열인 SREBP( 그림 3 )에 의해 전사적으로 제어됩니다 .
SREBP-1c는 편재적으로 발현되는 반면, SREBP-1a는 장 상피, 심장 및 대식세포에서 발현되고,
SREBP2는 간 및 지방 조직에서 발현된다.
SREBP1은 주로 FA 합성 유전자와 LDLR 의 발현을 조절하는 반면, SREBP2는 콜레스테롤 생합성 유전자 발현을 우선적으로 조절합니다.
비활성 SREBP는 ER 막에 있으며, 성숙한 SREBP는 핵으로 이동하고 표적 유전자 프로모터 내의 SRE 및 E-Boxes에 동종이량체로 결합합니다.
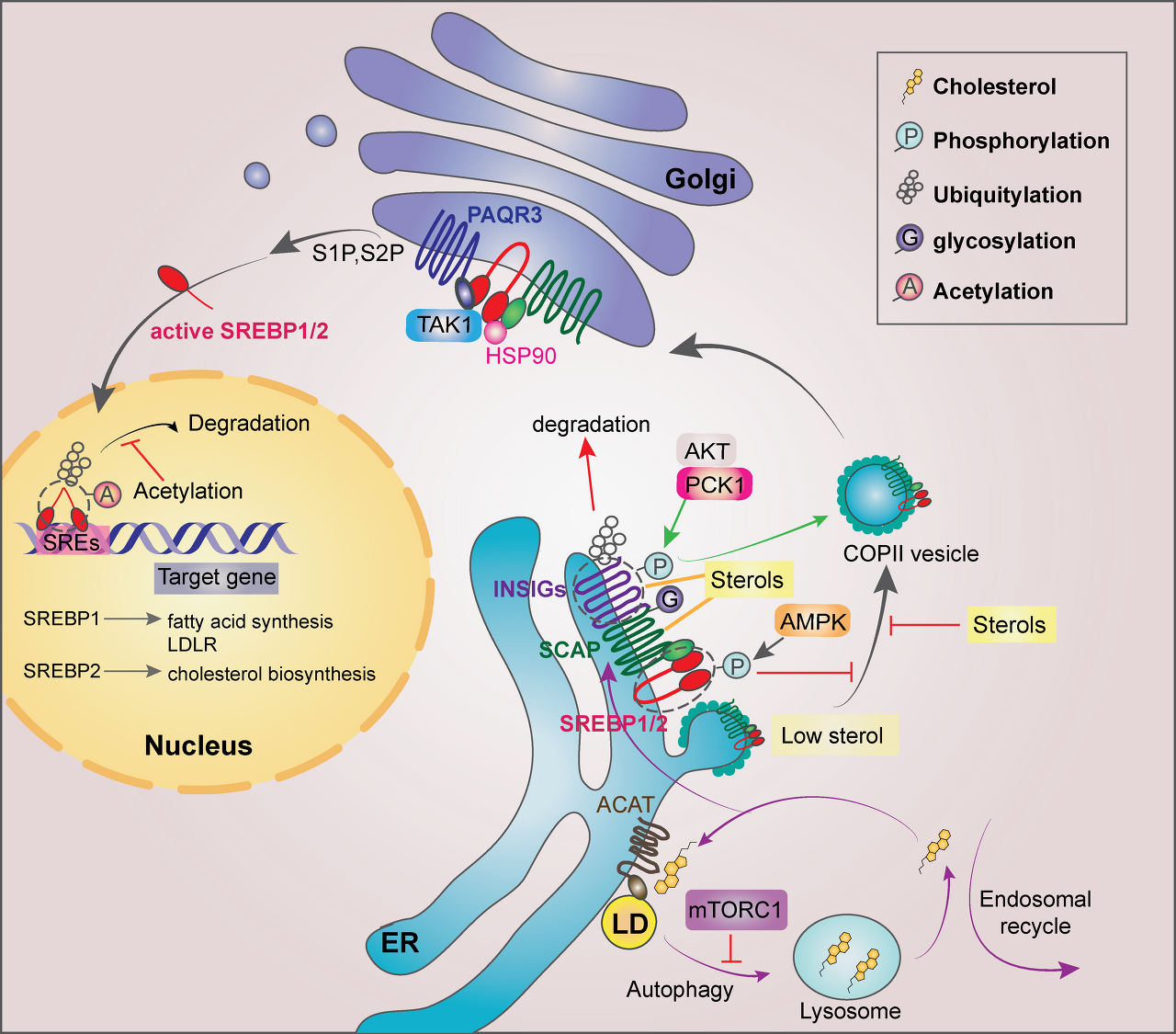
암세포에서 SREBP1/2의 조절..
SREBP 활동은 여러 수준과 다른 세포 내 위치에서 조절될 수 있습니다.
ER에서 스테롤은 SCAP에 결합하고 SREBP ER 출구를 위해 SCAP와 COPII 사이의 직접적인 상호작용을 방해합니다.
스테롤 수준이 감소하면 SCAP는 INSIG에서 해리되고 SCAP/SREBP가 COPII 코팅된 소포로 통합되는 것을 촉진합니다.
mTORC1은 자가포식과 리소좀에서 ER로의 콜레스테롤 수송을 억제하여 SREBP2 활성화를 유도합니다.
장쇄 불포화 FA는 INSIG1의 편재화 억제를 통해 SREBP 활성화를 억제합니다.
AKT 인산화된 PCK1은 INSIG1/2를 인산화하고 SREBP1/2 활성화를 위해 INSIG1/2에 대한 옥시스테롤의 결합을 방해합니다.
또한 활성화된 AMPK는 SREBP1/2를 ER에서 유지하기 위해 인산화할 수 있습니다.
EGFR 활성화는 SCAP의 N-글리코실화를 향상시키고, INSIG1에서 해리를 유발합니다.
골지체에서 SREBP1/2는 S1P와 S2P에 의해 절단되어 전사적으로 활성인 SREBP1/2를 방출합니다.
HSP90은 ER에서 Golgi로 SREBP-SCAP 복합 이동을 용이하게 합니다.
PAQR3는 골지체에서 SREBP 처리를 강화하는 반면, SREBP1/2의 TAK1 매개 인산화는 SREBP를 억제합니다.
핵에서 절단된 SREBP1/2는 표적 유전자의 프로모터 내 SRE에 결합합니다.
GSK3 인산화 SREBP1/2는 유비퀴틸화 및 분해를 겪으며, 이는 SREBP1/2의 유비퀴틸화 Lys 잔기의 아세틸화에 의해 상쇄될 수 있습니다.
반면 SREBP1/2의 TAK1 매개 인산화는 SREBP를 억제합니다.
핵에서 절단된 SREBP1/2는 표적 유전자의 프로모터 내 SRE에 결합합니다.
SREBP-1c는 편재적으로 발현되는 반면, SREBP-1a는 장 상피, 심장 및 대식세포에서 발현되고,
SREBP2는 간 및 지방 조직에서 발현된다.
SREBP1은 주로 FA 합성 유전자와 LDLR 의 발현을 조절하는 반면, SREBP2는 콜레스테롤 생합성 유전자 발현을 우선적으로 조절합니다.
비활성 SREBP는 ER 막에 있으며, 성숙한 SREBP는 핵으로 이동하고 표적 유전자 프로모터 내의 SRE 및 E-박스에 동종이량체로 결합합니다.
.SREBP 활성은 ER, Golgi 및 핵과 같은 여러 수준 및 다른 세포내 국소화에서 조절될 수 있습니다.ER에서 SREBP의 조절
SREBP 활성화는 ER의 스테롤 변동에 의해 조절됩니다.
콜레스테롤은 SCAP에 결합하여 SCAP와 COPII 간의 상호 작용을 방해하고 ER에서 SREBP를 유지합니다.
p53은 콜레스테롤 트랜스포터 유전자인 ATP-결합 카세트 트랜스포터(ABCA1)를 전사적으로 유도한다.
p53 손실 또는 ABCA1 제거는 원형질막에서 ER로 콜레스테롤의 역행 수송을 감소시켜 SREBP2 성숙 및 마우스 간 종양 형성을 촉진합니다.
ER에 상주하는 ACAT는 ER 콜레스테롤을 FA와 에스테르화하여 지질 방울에 저장하기 위한 콜레스테릴 에스테르를 형성하여 ER 콜레스테롤 수준을 제어합니다.
ACAT의 억제는 ER 콜레스테롤을 축적하고, 지질 방울 형성을 감소시키며, SREBP1 조절 유전자 발현을 차단함으로써 교모세포종 성장과 전립선암 세포의 공격성을 억제합니다.
mTORC1은 자가포식을 억제하고 원형질막으로의 엔도솜 재순환을 유지하여 막 유래 콜레스테롤이 리소좀에 도달하고 후속 ER 국소화를 방지하여 SREBP2 활성화를 유도합니다
골지체에서 SREBP의 조절
열 충격 단백질 90(HSP90)은 ER과 골지체 모두에서 SREBP-SCAP 복합체에 결합하여 이를 안정화시키고 골지로의 이동을 촉진하는 반면, HSP90 억제는 복합체의 프로테아좀 분해를 유도합니다.
핵 SREBP의 규제
mTORC1은 인산화 및 포스파티데이트 포스파타제 LPIN1의 세포질 보유를 통해 성숙한 SREBP1의 핵 국소화를 촉진하며, 이는 인산화되지 않은 상태에서 핵 주변부에서 SREBP1을 격리하여 억제합니다.
ER 스트레스 또는 포도당 결핍 시 ATF6은 SREBP2와 상호작용하여 HDAC1을 ATF6-SREBP2 복합체에 모집하고 SREBP2를 억제합니다
SREBP의 전사 조절과 지질 항상성에서 LXR의 역할
LXRα 및 LXRβ는 레티노이드 X 수용체와 이종이량체를 형성 하고 지질 합성을 촉진하기 위해 LXR 리간드로 작용하는 다른 핵 옥시스테롤의 존재 하에 SREBF1 프로모터의 보존된 LXR 반응 요소에 결합합니다.
암 세포에서 지질 대사에서 LXR의 역할은 여러 다운스트림 유전자의 발현으로 인해 복잡하며 LXR은 다양한 종양 세포의 성장 및 생존에 대해 양성 또는 음성 조절을 발휘하는 것으로 나타났습니다.
LXR은 상향 조절된 ABCA1 발현 에 의해 매개되는 콜레스테롤 유출 을 증가시키고 LDLR 분해를 위한 유비퀴틴 리가제인 LDLR(Idol)의 유도성 분해자의 발현 증가에 의해 매개되는 콜레스테롤 흡수를 감소시켜 세포내 콜레스테롤 수준을 감소시킬 수 있습니다.
대조적으로, SREBF1 을 활성화하는 것 외에도발현, LXR은 FASN 및 SCD1 발현을 유도하여 지방 생성을 증가시킬 수 있고 포스포프룩토키나제-2( PFK2 ) 및 글루코키나제(GCK1) 발현을 유도하여 해당과정을 향상시킬 수 있습니다.
LXR 활성화가 면역 세포 기능에 관여하는 세포 주기 조절자 및 유전자의 발현을 유도한다는 점을 감안할 때 온전한 종양 면역 미세 환경에서 암의 LXR을 연구하면 보다 통찰력 있는 정보를 해명할 수 있습니다.
지방분해 및 FAO
FAO는 미토콘드리아에서 장쇄 FA를 TCA 회로에 들어가는 아세틸-CoA로 전환하고, 전자 수송 사슬에 사용되는 조효소인 NADH와 FADH2로 전환합니다( 그림 4).).
FAO는 미토콘드리아에서 산화되기에는 FA 사슬이 너무 길어 퍼옥시좀에서 발생합니다.
외부 미토콘드리아 막에서 지방 아실-CoA는 CPT1에 의해 지방 아실카르니틴으로 전환되며, 이 동형 CPT1A, CPT1B 및 CPT1C는 각각 간, 근육 및 뇌에서 우세하게 발현됩니다.
미토콘드리아 내부 막에 위치한 카르니틴/아실카르니틴 트랜스로카제는 아실카르니틴을 미토콘드리아 기질로 이동시킵니다.
내막의 기질 쪽에 있는 CPT2는 acylcarnitine을 acyl-CoA로 재전환하여 아세틸-CoA로 절단되고 TCA 회로에 들어가 ATP와 말산 효소 의존적 NADPH를 생성합니다( 그림 4).).
또한, FAO 생성 시트레이트는 IDH 매개 이소시트레이트 산화에 의한 NADPH 생성을 위해 세포질로 내보낼 수 있습니다.
NADPH는 생합성 및 산화환원 항상성을 지원하는 환원제입니다.
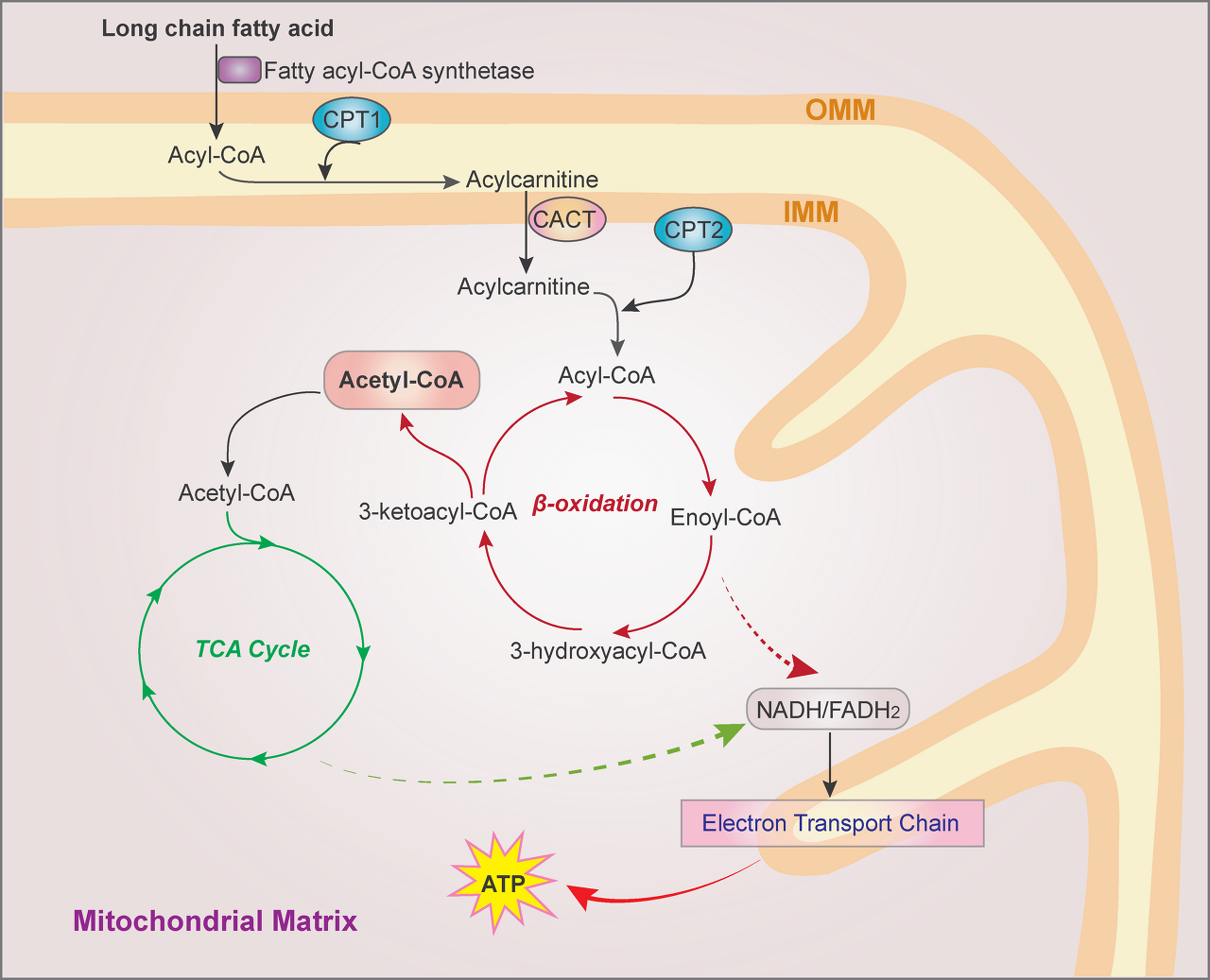
FAO 경로.
FA는 지방 아실-CoA 합성효소에 의해 지방 아실-CoA로 활성화됩니다.
외부 미토콘드리아 막에서 지방 아실-CoA는 CPT1에 의해 지방 아실카르니틴으로 전환되고 미토콘드리아 기질로 이동합니다.
그런 다음 내막의 기질 쪽에 있는 CPT2는 아실카르니틴을 아실-CoA로 재전환하고, 이는 아실-CoA 탈수소효소, 에노일-CoA 수화효소, 3-히드록시아실의 활성에 의해 순차적으로 촉매되는 반복된 4단계 주기에 의해 아세틸-CoA로 절단됩니다.
CoA 탈수소효소 및 3-케토아실-CoA 티올라제, 결과적으로 각 주기에서 2개의 탄소로 FA가 단축됩니다. 분해 생성물인 아세틸-CoA는 TCA 회로에 들어가고 생성된 NADH와 FADH2는 전자 수송 사슬에서 ATP를 생성하는 조효소로 사용됩니다.
CACT, carnitine/acylcarnitine translocase; IMM, inner mitochondrial membrane;
OMM, outer mitochondrial membrane.
FAO의 가장 두드러진 전사 조절자는 PPAR입니다.
삼중 음성 유방암에서 과발현되는 전골수구성 백혈병 단백질은 PGC-1α 아세틸화를 감소시켜 PPAR 및 FAO를 활성화합니다.
FAO는 AMPK 활성화에 의해 자극될 수 있습니다. AMPK는 또한 알로스테릭 CPT1 억제제인 ACC 생성 말로닐 CoA를 감소시킵니다.
인간 결장암에서 발현이 증가된 ACC1 유래 원형 RNA인 CircACC1은 대사 스트레스 하에서 AMPK 복합체의 조립 및 활성화를 조절하여 FAO 및 이종이식 종양 성장을 촉진합니다.
ATP 생산 및 세포 증식에서의 역할 외에도 FAO는 NADPH 항상성, 미토콘드리아 기능 및 세포 생존에 중요합니다.
FAO 억제는 상응하는 증가된 ROS 수준 및 신경교종 세포의 세포자멸사 비율과 함께 NADPH 수준을 감소시킵니다.
에너지 스트레스 동안 AMPK 활성화는 FAO를 통한 NADPH 생성을 증가시키고 FA 합성에서 NADPH 소비를 억제하여 폐암 세포 사멸을 억제합니다.
그러나 지나치게 활성화된 FAO는 세포독성을 유발할 수 있습니다. .
FAO의 역동적이고 균형 잡힌 제어는 필요한 ATP와 NADPH를 제공하고, 잠재적으로 독성이 있는 지질을 제거하고, 세포자멸사 경로를 억제하고, 동화작용에 필요한 대사 중간체를 생성함으로써 종양 세포 증식과 생존을 지원합니다.
암 치료를 위한 지질 대사 표적화
발암성 신호 전달과 지질 대사 사이의 통합 및 상호 조절은 암세포 성장, 생존, 증식, 이동, 침습 및 전이를 촉진합니다.
면역 세포, 지방 세포, 내피 세포 및 섬유 아세포를 포함한 종양 미세 환경의 암세포 및 기타 세포의 지질 대사는 동적으로 조절되고 상호 연결되며 다양한 수준에서 지질 대사에 개입하는 치료제를 개발하기 위해 많은 노력을 기울이고 있습니다.
콜레스테롤 합성을 표적으로 하기 위해 HMGCR 억제제와 같은 스타틴 계열 약물이 현재 여러 임상 시험에서 항암제로 테스트되고 있습니다.
일부 일치하지 않는 결과가 보고되었습니다. 후향적 연구에 따르면 스타틴 치료는 1차 화학요법과 함께 치료된 다발성 골수종, 결장직장암 및 전이성 췌장암 환자의 생존 기간을 연장했습니다.
대조적으로 SCLC, 전이성 결장직장암, 간세포암종 또는 위암 환자를 대상으로 한 무작위 3상 시험에서는 표준 화학요법에 프라바스타틴 또는 심바스타틴을 추가하는 것이 추가적인 이점을 제공하지 않는 것으로 나타났습니다(.
친유성 스타틴 약물은 더 쉽게 간외 세포로 들어가는 반면, 친수성 스타틴은 더 간에 선택적이라고 제안되었습니다.
또한 임상 데이터에 따르면 스타틴의 항암 효과는 용량과 시간에 모두 의존적입니다.
지질 흡수와 합성이 모두 암세포의 지질 자원에 기여한다는 점을 감안할 때 FA 또는 콜레스테롤 생합성을 억제하는 간단한 치료 전략은 식이 지질의 보상으로 인해 덜 효과적일 수 있습니다.
또한 동적 FAO 규제는 암 진행에 중요한 역할을 합니다.
'암치료' 카테고리의 다른 글
| 암의 지질 대사 변화: 예후 및 치료에 미치는 영향(허브 포함) (1) | 2022.09.30 |
|---|---|
| 암세포가 지질 대사를 리모델링하는 방법: 전사 인자를 표적으로 하는 전략 (0) | 2022.09.29 |
| 암의 지질 대사 환경과 새로운 치료적 관점 (0) | 2022.09.27 |
| 암 진행 및 치료 전략에서의 지질 대사 (프로토콜) (0) | 2022.09.26 |
| AMPK (0) | 2022.09.15 |